










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.30 no.3 Bogotá July/Sep. 2014
Caso Clínico
Variabilidad genotipo fenotipo en encefalopatía neurogastrointestinal mitocondrial MNGIE por una mutación sin codón de parada
Genotype phenotype variation in mitochondrial neurogatrointestinal encephalopathy MNGIE because of a mutation without stop codon
Blair Ortiz Giraldo (1), Diego Fernando Batero (2), Jorge Hernán Montoya (3), Juan Manuel Alfaro (4), Alejandra Wilches (5)
(1) Neurólogo infantil y pediatra. Grupo de Neurología infantil, Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia. Colombia
(2) Médico general de Neurología Integral de Caldas NIC y Hospital Departamental Santa Sofía de Caldas. Colombia
(3) Médico genetista del Hospital San Vicente Fundación Medellín. Colombia
(4) Endocrinólogo infantil y pediatra. Departamento de Pediatría, Facultad de Medicina. Universidad de Antioquia. Medellín, Colombia.
(5) Gastroenteróloga infantil y pediatra del Hospital San Vicente Fundación Medellín. Colombia.
Recibido: 28/01/14. Aceptado: 15/07/14.
Correspondencia: Blair Ortíz Giraldo: blairortiz@hotmail.com
RESUMEN
La encefalopatía neurogastrointestinal mitocondrial (MNGIE) es una enfermedad genética que se manifiesta desde los primeros años de vida con episodios de íleo, obstrucción intestinal, trastorno de deglución, falla de medro, miopatía, neuropatía periférica y leucoencefalopatía. Sin embargo, las manifestaciones clínicas pueden ser leves o incompletas. En la mayoría de los casos es producto de una mutación de novo, pero también puede heredarse de forma autosómica recesiva. Específicamente, la mutación c.1416 se asocia a MNGIE con neuropatía periférica severa. A continuación se describe el debut, características clínicas, hallazgos bioquímicos, neuroimagen y confirmación de la mutación c.1416 de un paciente con MNGIE pero sin enfermedad de motoneurona inferior.
PALABRAS CLAVE.: Mitocondrial, gastrointestinal, miopatía, encefalopatía, discapacidad cognitiva, neuropatía. (DECS).
SUMMARY
Mitochondrial neurogastrointestinal encephalopathy (MNGIE) is a genetic disease with onset from infancy and expressed with bowel obstruction, swallowing disorder, growth retardation, myopathy, peripheral neuropathy and cerebral leukoencephalopathy. The majority of cases are produced by a novo mutation and sometimes by an autosomic reccessive inheretance. MNGIE produced by c.1416 mutation has been associated with severe peripheral neuropathy. In the following report, we describe the onset disease, clinical features, biochemical data, cerebral magnetic resonance image, genetic test and literature review of a patient with MNGIE without inferior motoneuron disease and c.1416 mutation.
KEY WORDS. Mitochondrial, gastrointestinal, myopathy, encephalopathy, intellectual disability, neuropathy.(MeSH).
INTRODUCCIÓN
MNGIE es una enfermedad autosómica recesiva causada por mutaciones en el gen que codifica la enzima timidina fosforilasa (TP). El diagnóstico en los estadios iniciales es difícil pero posteriormente tiene manifestaciones clínicas más características y homogéneas. El diagnóstico definitivo recae en los estudios bioquímicos y genéticos (1). La enfermedad es infrecuente (2).
Se caracteriza por dismotilidad gastrointestinal severa, caquexia, oftalmoplejia progresiva externa, neuropatía periférica, leucoencefalopatia difusa y evidencia de disfunción mitocondrial (3). Las mutaciones con pérdida de función de la TP causan MNGIE. La TP cataliza el primer paso en la degradación de los nucleósidos pirimidínicos de timina (dThd) y deoxiuridina (dUrd) (4). La medición de la actividad de TP en leucocitos de pacientes con MNGIE ha revelado pérdida total o casi completa y acumulación plasmástica de dThd y dUrd (5).
Sin embargo, se han reportado pacientes con síntomas clínicos leves o incompletos de la enfermedad. La mutación c.1416 fue descrita por primera vez en el año 2011 en un paciente español con MNGIE y neuropatía severa (6). A continuación se presentan los aspectos clave para el diagnóstico clínico, de laboratorio y la confirmación genética de la mutación c.1416 en un paciente con MNGIE pero sin neuropatía periférica.
PRESENTACIÓN DE CASO
Paciente masculino de 16 años de edad que presenta desde el nacimiento debilidad para alimentarse, trastorno de deglución y constipación. A los 2 meses de vida inicia síndrome de pseudo-obstrucción intestinal, requiriendo manejo médico y quirúrgico en varias ocasiones pero sin encontrar patología (cirugías en blanco).
Tenía antecedentes personales de retardo del desarrollo motor; adquirió el sostén cefálico a los 6 meses, sedestación a los 8 meses y marcha a los 2 años; tuvo falla de medro. No tenía antecedentes familiares de enfermedades neurológicas ni neuromusculares y sus padres no eran consanguíneos.
A los 10 años requirió derivar la alimentación con gastrostomía y, más adelante, fundoplicatura de Nissen por íleo, bridas intestinales, enfermedad por reflujo gastroesofágico y vólvulo intestinal. Presentaba hiperlactatemia durante las intercurrencias infecciosas comunes (como resfriado común, diarrea y fiebre de origen viral) que lo llevaban a deterioro del estado de conciencia (somnolencia y obnubilación). Recibía hormona de crecimiento por síndrome de consunción por enfermedad crónica.
A los 11 años de vida estaba enflaquecido, tenía voz nasal, bradipsiquia, bradilalia, discapacidad cognitiva, hipotonía muscular, paresia leve (evaluación manual segmentaria de la fuerza de 4/5), fatiga muscular, hiporreflexia simétrica (1/4), respuesta plantar flexora y hepatomegalia leve (2cm debajo del reborde costal derecho con línea media clavicular). No tenía degeneración pigmentaria de la retina, ni esplenomegalia, ni alteraciones sensitivas y su desarrollo sexual era normal (Tanner III).
Por el curso de su enfermedad y los antecedentes personales se consideró una enfermedad metabólica con compromiso cerebral, muscular y gastrointestinal y se solicitaron los estudios que se muestran en la Tabla 1.
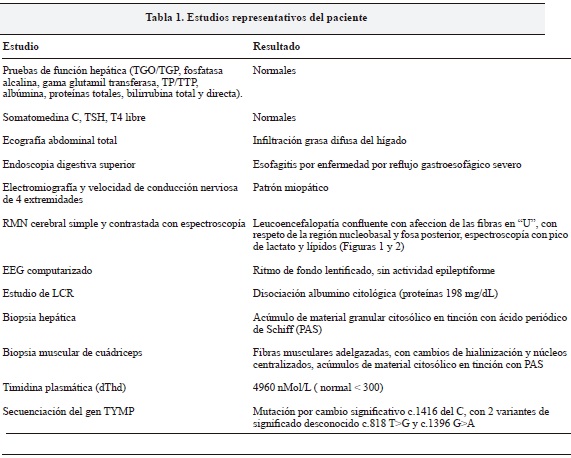
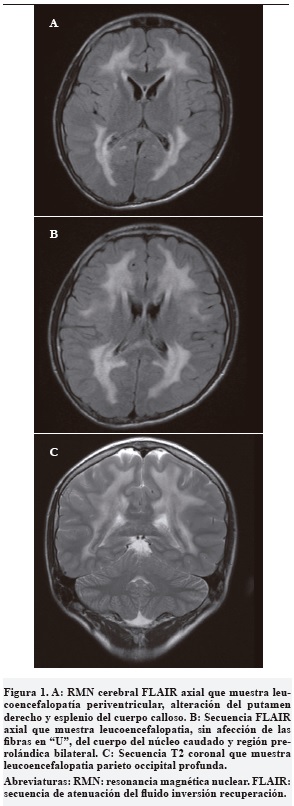
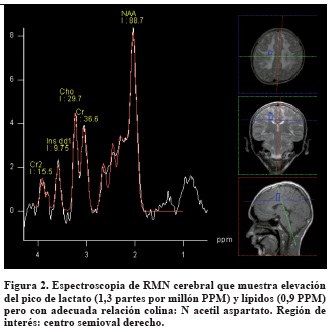
Con el diagnóstico de encefalomiopatía neurogastrointestinal mitocondrial se inició suplementación con fórmula polimérica y cofactores (multivitaminas con zinc, carbonato de calcio, calcitriol, D-ribosa y ubiquinol liposomal concentrado). Estos medicamentos fueron recibidos de forma inconstante por parte de su aseguradora de salud.
A los 16 años de vida presenta diarrea crónica, dolor abdominal intenso, deterioro del estado de conciencia y se documenta hipoglicemia. Es remitido a un hospital de mayor complejidad por emergencia abdominal. La gasimetría mostró acidemia metabólica persistente (pH 7,2, PCO2 35,3, PO2 45, HCO3 14, BE -11,5) e hiperlactacidemia (68 mmol/L). Se hace diagnóstico de abdomen agudo por peritonitis y el paciente es llevado a lavado peritoneal, donde se encuentra perforación intestinal. Luego, presenta deterioro hemodinámico, falla orgánica múltiple y fallece.
DISCUSIÓN
MNGIE es una enfermedad autosómica recesiva producida por depleción, deleción y mutaciones puntuales del DNA mitocondrial (7). La enfermedad fue originalmente descrita por Okamura y colaboradores como “miopatía oculoesquelética congénita con mitocondrias musculares y hepáticas anormales” (8). Posteriormente, la enfermedad se reportó bajo otras definiciones que incluían el síndrome POLIP: polineuropatía oftalmoplejía leucoencefalopatía intestinal pseudo-obstrucción (9), síndrome OGIMD: distrofia oculogastrointestinal muscular (10) y síndrome MEPOP: encefalomiopatía mitocondrial con polineuropatía sensorimotora, oftalmoplejía y pseudo-obstrucción (11).
Se puede distinguir clínicamente de otras encefalopatías por su presentación estereotipada. Sin embargo, sus manifestaciones se desarrollan durante la adolescencia y adultez, por lo que los métodos de tamización y diagnóstico definitivo son extremadamente útiles, especialmente en la fase temprana de la enfermedad.
Los pacientes pueden presentar oftalmoplejia progresiva externa y ptosis, semejante a otra enfermedad mitocondrial o miastenia gravis. Los que presentan neuropatía periférica desmielinizante pueden ser diagnosticados de polineuropatía desmielinizante inflamatoria crónica o de enfermedad de Charcot Marie Tooth (12). En nuestro caso clínico, la dismotilidad gastrointestinal fue la principal característica y pudo confundirse con síndrome de intestino irritable, enfermedad celiaca, síndrome de arteria mesentérica superior y anorexia nerviosa (13). El paciente que presentamos tuvo debut de la enfermedad desde el nacimiento con íleo, muchos episodios de pseudo-obstrucción intestinal, miopatía, discapacidad cognitiva y descompensación metabólica por acidemia láctica durante las intercurrencias infecciosas. Fue llevado en varias ocasiones a cirugías abdominales pero no se encontró patología inflamatoria.
En la mayoría de los pacientes con MNGIE hay anormalidades evidentes histológicamente en las mitocondrias de los músculos como fibras rojas rasgadas y fibras deficientes de citocromo C oxidasa. Genéticamente, se encuentran deleciones, depleciones y mutaciones puntuales somáticas en el músculo esquelético u otros tejidos afectados (14). En el paciente reportado se encontraron signos de miopatía crónica y acúmulos intracelulares de material PAS positivo en biopsia hepática, explicados por las alteraciones hepato-pancreáticas presentes en MNGIE.
La enfermedad se presenta por mutaciones en el gen nuclear del factor de crecimiento celular endotelial (ECGF1, por sus siglas en inglés), que codifica la TP (15). Este gen se localiza en el brazo largo del cromosoma 22 (22q13.33). Los pacientes tienen ausente la actividad TP y se acumula dThd y dUrd, lo que probablemente afecta la replicación y reparación del mtDNA (16). MNGIE es producida por la ausencia enzimática total o casi-total de TP. Los portadores asintomáticos tienen actividad TP parcial (alrededor del 35%). La TP convierte los dThd y dUrd a bases libres y deoxiribosa 1-fosfato. Mientras los niveles séricos de individuos sanos de los dos deoxinucleósidos son menores que 0,05 uM, en los pacientes con MNGIE se encuentran concentraciones entre 10 y 20 uM (17). Nuestro paciente tenía los niveles séricos dThd cien veces por encima del valor normal, lo que sugería una deficiencia de la TP.
Sólo los pacientes con MNGIE tienen elevación plasmática de dUrd (>5 umol/L) y dThd (>3 umol/L), mientras que los portadores de la mutación con actividad TP reducida no tienen nucleósidos de pirimidina detectables (<0,05 umol/L). Las pirimidinas son efectivamente catabolizadas en todos los portadores, incluyendo aquellos con actividad TP > 15% de controles sanos. Los pacientes con trastornos semejantes a MNGIE (MNGIE-like) no cumplen los criterios diagnósticos ni tienen nucleósidos de pirimidina detectables. La elevación plasmática de dThd y dUrd en MNGIE participan en la patogénesis de la enfermedad (18).
El análisis de la secuencia del gen TP permite identificar aproximadamente el 99% de las mutaciones patogénicas que ocurren dentro de las regiones codificantes del gen; sin embargo, esta estrategia tiene algunas limitaciones. El secuenciamiento de los exones que codifican proteínas puede obviar las mutaciones en el promotor, en los intrones no flanqueados y los polimorfismos recientemente identificados con implicaciones patológicas.
Hasta el momento se han reportado más de 60 mutaciones del gen TP en diversas poblaciones étnicas como judíos Ashkenazi y persas, Europa Oriental, Jamaica, Turquia, España, Francia, Tailandia, Irán y Japón (19-21). La mayoría son mutaciones sin sentido y menos frecuentemente microdeleciones, inserciones, mutaciones del sitio de corte y de duplicación. En nuestro paciente identificamos una mutación homocigota en el c.1416 del C, la cual ya había sido descrita por Torres y colaboradores, y que opera en los niveles translacionales o post-translacionales más que en la descomposición delectiva del mRNA de parada (6). Es de especial interés el hecho de que esta mutación haya sido asociada a neuropatía periférica severa y que nuestro paciente no tuviera enfermedad de motoneurona inferior.
La mejor opción terapéutica para MNGIE es el trasplante alogénico de células madre hematopoyéticas, porque restaura la actividad enzimática y reduce los niveles plasmáticos de dThd y dUrd (22, 23). Desafortunadamente, la mayoría de los pacientes no son candidatos por su pobre condición médica cuando se hace el diagnóstico definitivo.
La prevalencia de esta infrecuente y devastadora enfermedad pudo haber sido subestimada en el pasado, pero la identificación de la causa molecular de base y una mejor comprensión de las alteraciones bioquímicas han llevado a un diagnóstico más acertado. Finalmente, este reporte muestra que la mutación c.1416 del C del gen TP puede tener variantes en la presentación clínica de MNGIE y que es producto del defecto en la comunicación intergenómica del DNA nuclear y mitocondrial (24).
Conflicto de intereses.
Los autores declaran no tener conflicto de intereses.
REFERENCIAS
1. MARTÍ R, SPINAZZOLA A, TADESSE S, NISHINO I, NISHIGAKI Y, HIRANO M. Definitive Diagnosis of Mitochondrial Neurogastrointestinal Encephalomyopathy by Biochemical Assays. Clinical Chemistry 2004; 50 (1): 120-124. [ Links ]
2. NISHINO I, SPINAZZOLA A, PAPADIMITRIOU A, HAMMANS S, STEINER I, HAHN CD, ET AL. MNGIE: an autosomal recessive disorder due to thymidine phosphorylase mutations. Ann Neurol 2000; 47:792-800. [ Links ]
3. PAPADIMITRIOU A, COMI GP, HADJIGEORGIOU GM, BORDONI A, SCIACCO M, NAPOLI L, ET AL. Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology 1998; 51: 1086-92. [ Links ]
4. Desgranges C, Razaka G, Rabaud M, Bricaud H. Catabolism of thymidine in human blood platelets: purification and properties of thymidine phosphorylase. Biochim Biophys Acta 1981; 654: 211-8. [ Links ]
5. MARTÍ R, NISHIGAKI Y, HIRANO M. Elevated plasma deoxyuridine in patients with thymidine phosphorylase deficiency. Biochem Biophys Res Commun 2003; 303:14-8. [ Links ]
6. TORRES-TORRONTERAS J, RODRIGUEZ-PALMERO A, PINÓS T, ACCARINO A, ANDREU AL, PINTOS-MORELL G, MARTÍ R. A novel nonstop mutation in TYMP does not induce nonstop mRNA decay in a MNGIE patient with severe neuropathy. Hum Mutat. 2011; 32 (4): 2061-8. [ Links ]
7. NISHINO, I., SPINAZZOLA, A., AND HIRANO, M. Thymidine phosphorylase gene mutations in MNGIE, a human mitochondrial disorder. Science 1999; 283: 689-692. [ Links ]
8. OKAMURA K, SANTA T, NAGAE K, OMAE T. Congenital oculo-skeletal myopathy with abnormal muscle and liver mitochondria. J Neurol Sci 1976: 2779-91. [ Links ]
9. SIMON LT, HOROUPIAN DS, DORFMAN LJ, MARKS M, HERRICK MK, WASSERSTEIN P, ET AL. Polyneuropathy, ophthalmoplegia, leukoencephalopathy and intestinal pseudo-obstruction: POLIP syndrome. Ann Neurol 1990; 28 (3): 49-60. [ Links ]
10. IONASESCU VV. Oculogastrointestinal muscular dystrophy. Am J Med Genet 1983: 15 (1): 03-12. [ Links ]
11. ROWLAND LP. Progressive external ophthalmoplegia and ocular myopathies. In: VINKENS PJ, BRUYN GW, KLAWANS HL, editors. Handbook Clin Neurol, Vol. 62. Amsterdam: Elsevier science publishers; 1992: 287-329. [ Links ]
12. BEDLACK RS, VU TH, HAMMANS S, SPARR SA, MYERS B, MORGENLANDER J, ET AL. MNGIE neuropathy: 5 cases mimicking chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 2004; 29: 364-368. [ Links ]
13. HIRANO M, NISHIGAKI Y, MARTÍ R. MNGIE: a disease of two genomes. Neurologist 2004; 10 (1): 8-17. [ Links ]
14. PAPADIMITRIOU A, COMI GP, HADJIGEORGIOU GM, BORDONI A, SCIACCO M, NAPOLI L, ET AL. Partial depletion and multiple deletions of muscle mtDNA in familial MNGIE syndrome. Neurology 1998; 51 (4): 1086-92. [ Links ]
15. HIRANO M, MARTÍ R, VILA MR, NISHIGAKI Y. MtDNA maintenance and stability genes: MNGIE and mtDNA depletion syndromes. In: KOEHLER CM, BAUER MF, editors. Mitochondrial function and biogenesis. Berlin: Springer-Verlag; 2004: 177-200 (Topics in current genetics, vol. 8). [ Links ]
16. NISHIGAKI Y, MARTÍ R AND HIRANO M. ND5 is a hot-spot for multiple atypical mitochondrial DNA deletions in mitochondrial neurogastrointestinal encephalomyopathy. Hum Mo Genet 2004. 13: 91-101. [ Links ]
17. BJURSELL G AND REICHARD P. Effects of thymidine on deoxyribonucleoside triphosphate pools and deoxyribonucleic acid synthesis in Chinese hamster ovary cells. J Biol Chem 1973; 248: 3904-3909. [ Links ]
18. SPINAZZOLA A, MARTÍ R, NISHINO I, ANDREU A, NAINI A, TADESSE S, ET AL. Altered thymidine metabolism due to defects of thymidine phosphorylase. J Biol Chem 2002; 277: 4128-33. [ Links ]
19. GAMEZ J, LARA MC, MEARIN F, OLIVERAS-LEY C, RAGUER N, OLIVE M, ET AL. A novel thymidine phosphorylase mutation in a Spanish MNGIE patient. J Neurol Sci 2005; 228 (1): 35-9. [ Links ]
20. KINTARAK J, LIEWLUCK T, SANGRUCHI T, HIRANO M, KULKANTRAKORN K, MUENGTAWEEPONGSA S. A novel ECGF1 mutation in a Thai patient with mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Clinical Neurology and Neurosurgery 2007; 109: 613-616. [ Links ]
21. AYATOLLAHI P, TARAZI A, NAFISSI S. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Acta Medica Iranica 2006; 44 (2): 151-154. [ Links ]
22. HIRANO M, YEBENES J, JONES AC, NISHINO I, DIMAURO S, CARLO JR, ET AL. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) syndrome maps to chromosome 22q13.32-qter. Am J Hum Genet 1998; 63: 526-33. [ Links ]
23. SCHUPBACH WM, BENOIST JF, CASALI C, ELHASID R, FAY K, HAHN D, ET AL. Allogeneic hematopoetic stem cell transplantation (HSCT) for mitochondrial neurogastrointestinal encephalomyopathy (MNGIE). Neurology 2009; 73: 332. [ Links ]
24. SLAMA A, LACROIX C, PLANTE-BORDENEUVE V, LOMBES A, CONTI M, REIMUND JM, ET AL. Thymidine phosphorylase gene mutations in patients with mitochondrial neurogastrointestinal encephalomyopathy syndrome. Mol Genet Metab 2005; 84 (4): 326-31. [ Links ]
