










Serviços Personalizados
Journal
Artigo
 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkActa Neurológica Colombiana
versão impressa ISSN 0120-8748
Acta Neurol Colomb. vol.30 no.4 Bogotá out./dez. 2014
Caso clínico
Neuropatía hereditaria con susceptibilidad a la parálisis por presión. Estudio clínico, neurofisiológico y genético de 3 casos esporádicos
Hereditary neuropathy with liability to pressure palsy. Clinical, neurophysiological and genetic study of 3 sporadic cases
Antonio Díaz Negrillo (1).
(1) Departamento de Neurofisiología Clínica. Hospital Universitario Infanta Elena. Madrid. España.
Recibido: 3/04/14. Aceptado: 3/10/14.
Correspondencia: Antonio Díaz Negrillo: antoniodnegrillo@yahoo.es
RESUMEN
Introducción. La neuropatía hereditaria con susceptibilidad a la parálisis por presión (NHPP) es una enfermedad genética que afecta fundamentalmente al componente mielínico de los nervios periféricos. Este estudio pretende describir detalladamente tres casos no emparentados familiarmente, así como realizar una revisión bibliográfica actualizada sobre el tema.
Casosclínicos. Pacientes de 23, 42 y 41 años estudiados por sospecha de neuropatía cubital (casos 1 y 2) y de síndrome del túnel carpiano bilateral (caso 3).
Resultados. Los estudios neurofisiológicos mostraron la existencia de una polineuropatía sensitivo-motora de predominio desmielinizante y mayor intensidad en localizaciones susceptibles al atrapamiento nervioso. El estudio genético confirmó en todos ellos la existencia de una deleción a nivel del gen PMP22 (cromosoma 17p11.2).
Conclusiones. Esta neuropatía hereditaria puede simular una simple neuropatía compresiva, estando por ello infradiagnosticada. Una anamnesis completa, así como un riguroso estudio neurofisiológico son fundamentales para una orientación diagnóstica adecuada.
PALABRAS CLAVE. Parálisis por presión, Neuropatía hereditaria, Neuropatía tomacular, Polineuropatía, Desmielinización, Electromiografía (DECS).
SUMMARY
Introduction. Hereditary neuropathy with liability to pressure palsy (HNPP) is a genetic disease that primarily affects the myelin of peripheral nerves. This study aims to describe in detail three cases with no familiar blood-ties and do an updated literature review on the topic.
Clinical cases. 23, 42 and 41 years old patients studied for suspected ulnar neuropathy (cases 1 and 2) and bilateral carpal tunnel (case 3) syndrome.
Results. The electromyographic examination revealed the existence of a sensory-motor demyelinating polyneuropathy of greater intensity in locations susceptible to nerve entrapment. The genetic study confirmed in all patients the existence of a deletion at the level of PMP22 gene (chromosome 17p11.2).
Conclusions. This hereditary neuropathy can simulate a simple compressive neuropathy. Therefore it is underdiagnosed. A thorough anamnesis and a rigorous neurophysiological study are essential in a proper diagnostic orientation .
KEY WORDS. Pressure Palsy, Hereditary Neuropathy, Tomacular Neuropathy, Polineuropathy, Demyelination, Electromyography (MeSH)..
INTRODUCCIÓN
La neuropatía hereditaria con predisposición a la parálisis por presión (NHPP) se caracteriza por una tendencia a desarrollar episodios de parálisis periférica, recidivantes e indoloras provocadas por traumatismos de baja intensidad o procesos compresivos (1, 2). La mayor parte de los casos están asociados con la deleción de la región 17p11.2 del gen que codifica la creación de una proteína periférica de la mielina (PMP22) heredándose de forma autosómica dominante (3). Las primeras descripciones de esta patología fueron hechas por Jong en 1947, quien publicó el caso de una familia afectada por un proceso de parálisis recurrente con carácter hereditario (4). Recientemente se ha publicado un nuevo caso perteneciente a dicha familia (5). Esta enfermedad aparece habitualmente en la adolescencia o el periodo de adulto joven. La presentación clínica es muy variada, aunque generalmente suelen afectarse los nervios mediano y cubital en los miembros superiores y el nervio peroneal en los miembros inferiores. Las áreas de lesión suelen ser lugares anatómicos en los que el nervio es más vulnerable y presenta propensión a la compresión (túnel carpiano, canal epitrócleo-olecraniano, cabeza del peroné etc) (6). La forma de presentación clásica es la mononeuropatía, si bien a nivel neurofisiológico son más frecuentes como formas de presentación la polineuropatía sensitivo-motora desmielinizante y la mononeuropatía múltiple o incluso la plexopatía (7, 8). La recuperación clínica suele ser habitual en el curso evolutivo de la enfermedad; no obstante, las recurrencias son características. El diagnóstico de sospecha se fundamenta en la historia clínica y los hallazgos neurofisiológicos, mientras que el diagnóstico de confirmación se logra con el estudio genético y la biopsia neural (9).
Presentamos el caso de tres pacientes no emparentados familiarmente que han sido diagnosticados de NHPP al ser estudiados en la Unidad de Neurofisiología Clínica por sospecha de mononeuropatías compresivas.
PRESENTACIÓN DEL CASO
Caso 1.
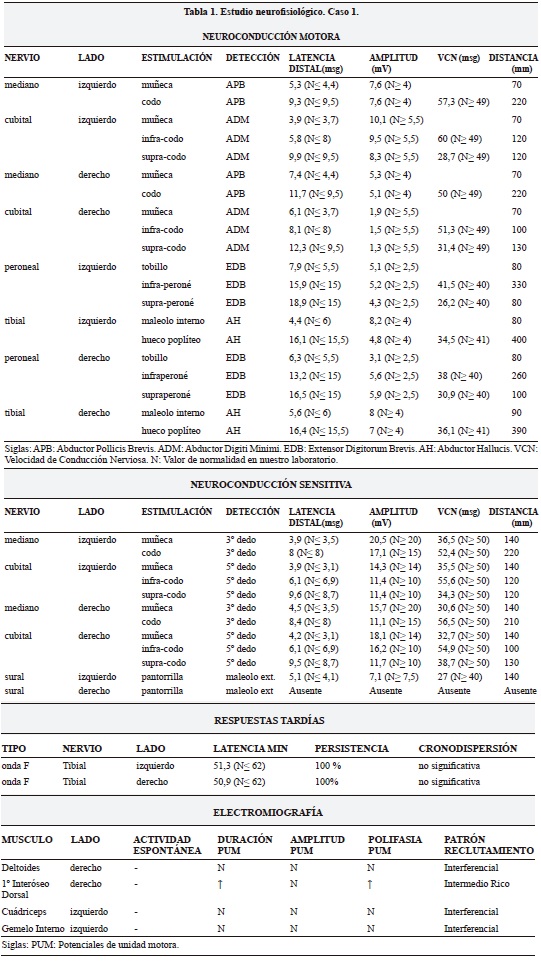
Varón de 23 años de edad, sin antecedentes médicos de interés, estudiado por cuadro de hipoestesia en el territorio cubital de la mano derecha de una semana de evolución. En la exploración física se destaca como único hallazgo un adormecimiento del borde cubital del 4º dedo y de todo el 5º dedo de la mano derecha. El paciente refirió haber presentado episodios similares, aunque menos intensos. Mencionó también haber presentado en alguna ocasión episodios de hipoestesia en cara antero-externa de la pierna izquierda y en cara dorsal del pie izquierdo con ligero déficit para la flexión dorsal del mismo, de varios días de duración, con mejoría espontánea. Se le realizó un estudio analítico (perfil neurológico) compuesto por hemograma y bioquímica de sangre y orina, proteinograma, hormonas tiroideas, vitamina B12 y ácido fólico, hemoglobina glicosilada, serologías (VIH, borrelia), pruebas treponémicas (ELISA) así como estudio de anticuerpos anti nucleares, anticuerpos anti citoplasma de neutrófilo y factor reumatoide. Todos los resultados resultaron normales. Se realizó un electroneuromiograma en el que se estudió la conducción de ambos nervios medianos, cubitales, peroneales profundos, tibiales (con ondas F) y surales, así como la actividad electromiográfica de los músculos deltoides y primer interóseo dorsal derechos, y cuadriceps y gemelo interno izquierdos. El paciente mide 1,75 metros y las mediciones se realizaron a una temperatura ambiental de 20ºC.
Los hallazgos neurofisiológicos (Tabla 1) revelaron la existencia de una polineuropatía sensitivo-motora de predominio desmielinizante, con afectación de ambos miembros superiores e inferiores y más acusada en los lugares de compresión nerviosa (canal epitrócleo-olecraniano, cabeza del peroné). Ante la sospecha diagnóstica de una posible neuropatía tomacular se decidió realizar un estudio genético con base en el siguiente procedimiento: extracción de ADN (Bio robot EZ1 Qiagen). Purificación del ADN contenido en las células de sangre periférica, reacción en Cadena de la Polimerasa de la región de interés, aplicación de la técnica MLPA (Multiplex Ligation-dependent Probe Amplification) usando kit MRC Holland específico para PMP22, uso de control ADN de individuo sano e interpretación de la altura de los picos observados. Se realizó, además, un estudio de microsatélites o STR (Short Tandem Repeat). Los resultados del estudio genético pusieron de manifiesto la existencia de una deleción a nivel del gen PMP 22. Este dato confirmaba el diagnóstico de sospecha.
Caso 2.
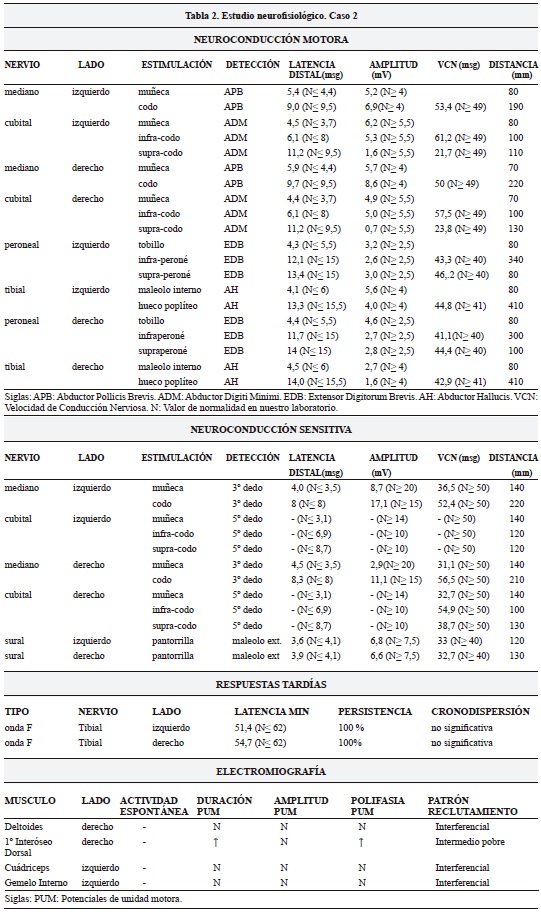
Mujer de 42 años de edad, sin antecedentes médicos de interés, estudiada por parestesias en la cara cubital del antebrazo derecho y en los dedos 4º y 5º de la mano derecha desde hace aproximadamente tres años. La exploración física reveló una hipoestesia táctil y termoalgésica de los dedos 4º (borde cubital) y 5º de mano derecha, así como debilidad en la abducción del 5º dedo de la mano derecha. Se le realizaron una analítica completa y un estudio neurofisiológico (similar al caso 1). La paciente mide 1,69 metros y las mediciones se realizaron a una temperatura ambiental de 20ºC. La analítica resultó normal y los hallazgos neurofisiológicos (Tabla 2) evidenciaron una polineuropatía sensitivo-motora localizada fundamentalmente en ambos miembros superiores, de predominio desmielinizante y más acusada en los lugares de compresión nerviosa (túnel carpiano y canal epitrócleo-olecraniano). Ante la sospecha clínica y diagnóstica de una posible neuropatía tomacular se decidió realizar un estudio genético aplicando la metodología descrita en el caso 1. Los resultados del estudio genético demostraron la existencia de una deleción a nivel del gen PMP 22, lo que confirmaba el diagnóstico de sospecha.
Caso 3.
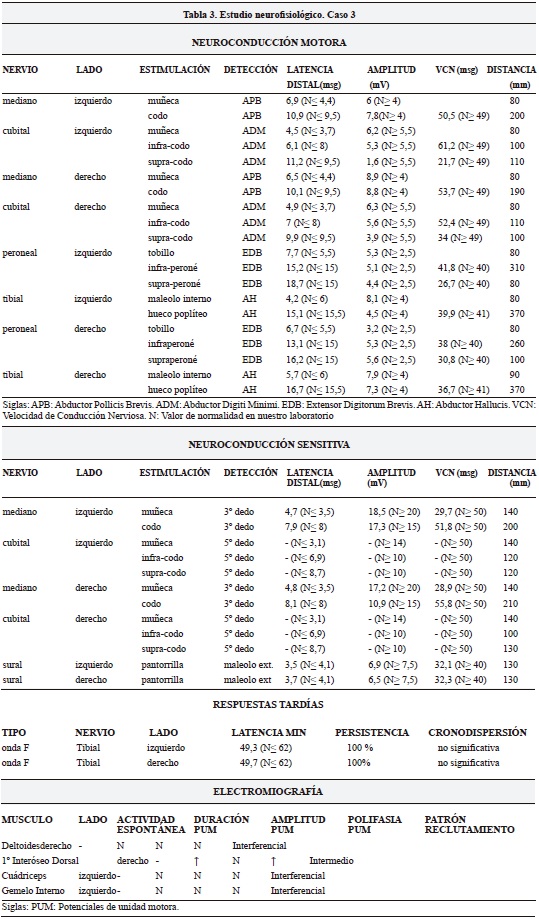
Mujer de 41 años de edad, sin antecedentes de interés, en estudio por un posible síndrome del túnel carpiano bilateral de varios años de evolución. En la exploración física se constató un test de Phalen y una maniobra de Tinel positivos en ambas manos. La paciente comentó que en anteriores ocasiones había notado parestesias persistentes en cara antero-externa de ambas piernas al cruzarlas una sobre la otra y que ella atribuyó a un problema postural. Se le realizó un estudio analítico y neurofisiológico similar al descrito en el paciente 1. La paciente mide 1,65 metros y las mediciones se realizaron a una temperatura ambiental de 20ºC. El estudio analítico fue normal. El estudio neurofisiológico (Tabla 3) evidenció en este caso también una polineuropatía sensitivo-motora de predominio desmielinizante y más acusada en los lugares de compresión nerviosa (túnel carpiano y canal epitrócleo-olecraniano, cabeza del peroné, etc.). Ante la sospecha clínica y diagnóstica de una posible neuropatía tomacular se decidió realizar un estudio genético aplicando la metodología descrita en el caso 1 y confirmando la existencia de una deleción a nivel del gen PMP 22.
DISCUSIÓN
La neuropatía hereditaria con predisposición a la parálisis por presión es una enfermedad neurológica infrecuente, de diagnóstico complejo y que, en ocasiones, puede simular otras neuropatías más frecuentes, como sucede con los casos que presentamos. La mayor parte de los pacientes presenta una deleción de 1,5 Mb en la región 17p11.2 del gen que codifica la proteína periférica de la mielina (PMP22), pero existen también casos en los que se dan mutaciones puntuales de la proteína PMP22, aunque con signos clínicos similares o más leves (9, 10). También se han publicado casos de pacientes en los que esta alteración genética está ausente, que están asintomáticos (11), o que presentan características clínicas similares con otros genes implicados (12). Esta enfermedad está emparentada con el síndrome de Charcot-Marie-Tooth. En el síndrome de Charcot-Marie-Tooth, se altera la misma región del gen, produciéndose, en este caso, una duplicación, y sugiriendo que ambas enfermedades pueden ser el resultado de un cruzamiento desigual en el proceso de meiosis.
En la neuropatía hereditaria con predisposición a la parálisis por presión, el cuadro clínico suele debutar en el adulto joven aunque existen casos descritos en la infancia (13, 14) y en personas ancianas (15). Es más frecuente en varones (aunque en nuestra muestra predomina el sexo femenino) (16). A pesar de presentar una herencia autosómica dominante, cerca del 40% de los pacientes representan casos aislados, como son los que presentamos. Este porcentaje de pacientes supone un diagnóstico más complejo dada la baja sospecha clínica. En esta línea, Beydoun y cols. reportaron en 2008 siete pacientes sin historia familiar conocida de NHPP y con diagnóstico genético confirmatorio (17).
La forma de presentación de la enfermedad suele incluir déficits sensitivo-motores asociados, en los que el nervio cubital es el más frecuentemente afectado (como en los casos 1 y 2 que presentamos). No obstante, existen pocos trabajos que documenten la afectación simultánea de varios nervios periféricos en diferentes partes de su trayecto. Farooq y cols. reportaron en 2008 el estudio de un paciente de 42 años que presentaba cinco procesos compresivos en cuatro nervios diferentes indicando que, desde su conocimiento, era el primer caso publicado con ese número de neuropatías compresivas (18). Los casos que actualmente presentamos superan este número de procesos compresivos simultáneos. No nos consta que se hayan publicado referencias concretas sobre la coexistencia de un número tan elevado de procesos compresivos simultáneos. Los hallazgos neurofisiológicos encontrados en nuestros casos ponen de manifiesto la existencia de una polineuropatía con afectación sensitiva y motora, de carácter desmielinizante, distribución bilateral, afectación de miembros inferiores y superiores y más acusada en estructuras anatómicas susceptibles de compresión nerviosa. En los casos 1 y 3 la afectación comprende a ambos nervios medianos, cubitales, peroneales y surales. En el caso 2, sin embargo, los nervios afectados son ambos medianos, cubitales y surales (encontrándose, por el momento, ambos nervios peroneales y tibiales posteriores conservados). En términos diagnósticos, Meier y cols. propusieron en 1982 los siguientes criterios diagnósticos: 1) herencia autosómica dominante, 2) clínica de mononeuropatía simple o múltiple recurrente y frecuentemente relacionada con traumatismos, 3) disminución significativa de la velocidad de conducción nerviosa sensitiva y motora en los nervios comprometidos y en los no afectados, 4) hallazgos histopatológicos característicos en la biopsia del nervio sural, con engrosamientos mielínicos focales (tomáculas) y desmielinización segmentaria (19). A su vez, Andersson y cols. han planteado como método para evaluar el grado de desmielinización distal el llamado Índice de Latencia Terminal, que se obtiene al dividir la distancia terminal (en milímetros) entre el producto de la latencia motora distal (en milisegundos) por la velocidad de conducción (metros/segundos), e indicando que este parámetro es menor en la neuropatía hereditaria con predisposición a la parálisis por presión que en otras polineuropatías más comunes como la diabética (20).
Podemos señalar que actualmente, con la sensibilidad y especificidad que aportan los estudios genéticos (21), estos criterios podrían ser modificables de tal forma que no sea estrictamente necesaria la biopsia del nervio sural para llegar a un diagnóstico de confirmación. En los tres casos que presentamos ha sido posible realizar un diagnóstico genético de confirmación partiendo de una anamnesis dirigida y un riguroso estudio neurofisiológico. Otro aspecto importante a tener en cuenta es la necesidad de realizar un minucioso diagnóstico diferencial con patologías que pueden presentarse clínicamente de forma parecida. Entre ellas están las neuropatías compresivas de origen diferente al que nos ocupa (síndrome del túnel carpiano), así como otras neuropatías hereditarias (Charcot-Marie-Tooth), plexopatías o incluso polineuropatías adquiridas de origen tóxico (alcohol), metabólico (diabetes mellitus, uremia) o incluso autoinmune (polineuropatía desmielinizante inflamatoria aguda o crónica) (22, 23). En esta línea es muy importante la realización de un completo estudio de laboratorio.
Con este trabajo pretendemos poner de manifiesto la importancia de realizar un examen neurofisiológico completo para poder llevar a cabo una aproximación diagnóstica correcta. Esta idea es compartida por otros autores (24). Es recomendable aplicar un protocolo electrodiagnóstico a aquellos pacientes con sospecha clínica fundada, que comprenda el estudio electroneurográfico con valoración de la velocidad de conducción nerviosa proximal y distal en nervios motores y sensitivos de las cuatro extremidades. Esto, además de estudiar las respuestas tardías (ondas F) de al menos dos nervios y completar la exploración con un estudio electromiográfico de la musculatura proximal y distal de al menos dos miembros. Ello permitirá, como se muestra en este trabajo, minimizar el número de falsos negativos optimizando por tanto la rentabilidad diagnóstica de la exploración.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
REFERENCIAS
1. BEHSE F, BUCHTHAL F, CARLSEN F, KNAPPEIS GG. Hereditary neuropathy with liability to pressure palsies. Electrophysiological and histopathological aspects. Brain 1972; 95:777-794. [ Links ]
2. RANA AQ, MASROOR MS. Hereditary neuropathy with liability to pressure palsy: a brief review with a case report. Int J Neurosci. 2012, Mar; 122(3):119-123. [ Links ]
3. CHANCE PF, ALDERSON MK, LEPPIG KA, LENSCH MW, MATSUNAMI N, SMITH B ET AL. DNA deletion associated with hereditary neuropathy with liability to pressure palsies. Cell 1993; 72:143-151. [ Links ]
4. KOEHLER PJ. Hereditary neuropathy with liability to pressure palsies: the first publication (1947). Neurology 2003; 60:1211-1213. [ Links ]
5. KOEHLER PJ, BAAS F. Hereditary neuropathy with liability to pressure palsies. Diagnosis in the first family (1947) confirmed. J Peripher Nerv Syst. 2012, Dec; 17(4):412-3. [ Links ]
6. DEL COLLE R, FABRIZI GM, TURAZZINI M, CAVALLARO T, SILVESTRI M, RIZZUTO N. Hereditary neuropathy with liability to pressure palsies: electrophysiological and genetic study of a family with carpal tunnel syndrome as only clinical manifestation. Neurol Sci 2003; 24:57-60. [ Links ]
7. MOUTON P, TARDIEU S, GOUIDER R, BIROUK N, MAISONOBE T, DUBOURG O, ET AL. Spectrum of clinical and electrophysiologic features in HNPP patients with the 17p11.2 deletion. Neurology 1999; 22;52:1440-1446. [ Links ]
8. MARTINELLI P, FABBRI R, MORETTO G, GABELLINI AS, D'ALESSANDRO R, RIZZUTO N. Recurrent familial brachial plexus palsies as the only clinical expression of tomaculous neuropathy. Eur Neurol 1989; 29:61-66. [ Links ]
9. PAPROCKA J, KAJOR M, JAMROZ E, JEZELA-STANEK A, SEEMAN P, MARSZAL E. Hereditary neuropathy with liability to pressure palsy. Folia Neuropathol 2006; 44:290-294. [ Links ]
10. CASASNOVAS C, BANCHS I, DE JORGE L, ANTÓNIA ALBERTÍ M, MARTÍNEZ-CAMPO Y, POVEDANO M. ET AL. A novel small deletion in PMP22 causes a mild hereditary neuropathy with liability to pressure palsies phenotype. Muscle Nerve. 2012, Jan; 45(1):135-138. [ Links ]
11. LUIGETTI M, CONTE A, MADIA F, MEREU ML, ZOLLINO M, MARANGI G, ET AL. A new single-nucleotide deletion of PMP22 in an HNPP family without recurrent palsies. Muscle Nerve 2008; 38:1060-1064. [ Links ]
12. MAGOT A, LATOUR P, MUSSINI JM, MOURTADA R, GUIHENEUC P, PEREON Y. A new MPZ mutation associated with a mild CMT1 phenotype presenting with recurrent nerve compression. Muscle Nerve 2008; 38: 1055-1059. [ Links ]
13. EIRÍS-PUÑAL J, VIDAL-LIJÓ M, BARROS-ANGUEIRA F, LÓPEZ-FERNÁNDEZ MJ, PINTOS-MARTÍNEZ E, BEIRAS-IGLESIAS A ET AL. Neuropatía hereditaria con parálisis sensible a la presión (neuropatía tomacular). Estudio clínico, electrofisiológico y molecular de dos familias afectadas. Rev Neurol 2000; 31:506-510. [ Links ]
14. FLOR-DE-LIMA F, MACEDO L, TAIPA R, MELO-PIRES M, RODRIGUES ML. Hereditary neuropathy with liability to pressure palsy: a recurrent and bilateral foot drop case report. Case Rep Pediatr. 2013; 2013: 230541. doi: 10.1155/2013/230541. Epub 2013 Oct 23. [ Links ]
15. KAWAGUCHI N, SUZUKI N, TATEYAMA M, TAKAI Y, MISU T, NAKASHIMA I, ET AL. Two cases of elderly-onset hereditary neuropathy with liability to pressure palsy manifesting bilateral peroneal nerve palsies. Case Rep Neurol. 2012, Sep; 4(3):149-155. [ Links ]
16. MANGANELLI F, PISCIOTTA C, DUBBIOSO R, MARUOTTI V, IODICE R, NOTTURNO F, ET AL. Electrophysiological comparison between males and females in HNPP. Neurol Sci. 2013, Aug; 34(8):1429-1432. [ Links ]
17. BEYDOUN SR, SYKES SN, GANGULY G, LEE TS. Hereditary neuropathy with liability to pressure palsies: description of seven patients without known family history. Acta Neurol Scand 2008;117:266-272. [ Links ]
18. FAROOQ MU, MARTIN JH, ANDARY MT. Unusual presentation of hereditary neuropathy with liability to pressure palsies. J Brachial Plex Peripher Nerve Inj. 2008, Jan, 24;3:2. [ Links ]
19. MEIER C, MOLL C. Hereditary neuropathy with liability to pressure palsies: report of two families and review of the literature. J Neurol. 1982; 228:73-94. [ Links ]
20. ANDERSSON PB, YUEN E, PARKO K, SO YT. Electrodiagnostic features of hereditary neuropathy with liability to pressure palsies. Neurology 2000;54:40-44. [ Links ]
21. STANGLER HERODEZ S, ZAGRADISNIK B, ERJAVEC SKERGET A, ZAGORAC A, KOKALJ VOKAC N. Molecular diagnosis of PMP22 gene duplications and deletions: comparison of different methods. J Int Med Res 2009; 37:1626-1631. [ Links ]
22. STANTON M, PANNONI V, LEWIS RA, LOGIGIAN EL, NAQUIB D, SHY ME, ET AL. Dispersion of compound muscle action potential in hereditary neuropathies and chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 2006;34:417-422. [ Links ]
23. HUGHES R, BEVILACQUA J. Neuropatía hereditaria con susceptibilidad a la parálisis por compresión. Presentación de casos clínicos y electrofisiológicos. Rev Hosp Clín Univ Chile 2009; 20:189-193. [ Links ]
24. LUIGETTI M, DEL GRANDE A, CONTE A, LO MONACO M, BISOGNI G, ROMANO A ET. AL. Clinical, neurophysiological and pathological findings of HNPP patients with 17p12 deletion: a single-centre experience. J Neurol Sci. 2014. Jun 15; 341(1-2): 46-50. [ Links ]
