










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkActa Neurológica Colombiana
versión impresa ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.1 Bogotá ene./mar. 2015
https://doi.org/10.22379/242240222
Trabajo original
Características clínicas y electroencefalográficas de los pacientes con Síndrome de Lennox-Gastaut en el programa de epilepsia de la U. Antioquia. Medellín 2007 - 2012.
Clinical and electroencephalographic characteristics of patients with Lennox-Gastaut Syndrome within the epilepsy program attended at Antioquia University. Medellin 2007 - 2012
Fabián Leonardo Fernández Echávez (1), Carolina Serrano Tabares (2), Rodrigo Andrés Solarte Mila (3), José William Cornejo Ochoa (4)
(1) Pediatra. Fellow de Neurología Infantil II Año, Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia. Investigador Principal.
(2) Pediatra. Fellow de Neurología Infantil II Año, Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia. Coinvestigadora.
(3) Neurólogo Clínico. Epileptólogo. Profesor Neurología Infantil, Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia. Coinvestigador.
(4) Neurólogo Infantil. Msc en Epidemiología. Profesor Neurología Infantil, Departamento de Pediatría, Facultad de Medicina, Universidad de Antioquia. Coinvestigador.
Recibido: 5/03/13. Aceptado: 20/01/15.
Correspondencia: Fabián Leonardo Fernández Echávez: fleofdez@hotmail.com
Resumen
Objetivo: describir las características clínicas y electroencefalográficas en una muestra de pacientes con síndrome de Lennox-Gastaut diagnosticados en el programa de epilepsia de la Universidad de Antioquia en Medellín entre 2007 y 2012.
Materiales y métodos: se trata de un estudio observacional, descriptivo y retrospectivo. La población de estudio estuvo conformada por todos los registros de pacientes con diagnóstico de síndrome de Lennox-Gastaut incluidos en la base de datos del programa de epilepsia de la Universidad de Antioquia y que fueron evaluados por videomonitoreo electroencefalográfico. Las variables clínicas y electroencefalográficas fueron examinadas. Para el análisis se utilizó el programa estadístico SPSS.
Resultados: se revisaron 18 videotelemetrías. El promedio de edad fue 19,89 años, con igualdad de género en un 50%. La mitad de los pacientes presentaba antecedentes perinatales de riesgo. La edad promedio de la primera crisis fue de 4,67 años y el número promedio de crisis por semana fue de 31,17. Las crisis más frecuentes fueron las ausencias atípicas en 17 pacientes (94,4%). El medicamento más utilizado fue el ácido valproico. En todos los pacientes se encontró retardo mental y los hallazgos electroencefalográficos característicos del síndrome, tanto en vigilia como en sueño. En el sueño superficial se observó la mayoría de anormalidades.
Conclusiones: el síndrome de Lennox-Gastaut es una de las encefalopatías epilépticas más severas de inicio en la niñez, conlleva grandes costos sociales y económicos y tiene un pobre pronóstico debido a sus condiciones mórbidas asociadas.
Palabra clave: Antiepilépticos. Crisis. Elecroencefalografía. Epilepsia. Retardo mental. Síndrome de Lennox-Gastaut (DeCS).
Summary
Objetive: To describe the clinical and electroencephalographic features in a sample of patients diagnosed with Lennox-Gastaut syndrome. The patients were part of the epilepsy program at the University of Antioquia in Medellin between 2007 and 2012.
Materials and methods: This was completed with an observational, descriptive and retrospective method. The data used was taken from the records of all patients diagnosed with Lennox-Gastaut syndrome included in the epilepsy program at the University of Antioquia and who were evaluated by EEG video monitoring. Clinical and electroencephalographic variables were analyzed. For the analysis we used SPSS.
Resualts: We reviewed 18 video EEG. The average age of the patients was 19,89 years, with the gender being equally being balanced. Half of the patients had a prenatal risk. The average age of the first seizure was at 4,67 years and the average number of attacks per week was 31,17. The most frequent were atypical absence seizures in 17 patients (94,4%). The most commonly used drug was valproic acid. All patients experienced mental retardation and characteristic electroencephalographic findings of the syndrome, during both times of wakefulness and sleep. Most abnormalities were observed during superficial sleep.
Conclusions: The Lennox-Gastaut syndrome is one of the most severe epileptic encephalopathies with onset during childhood and with large social and economic costs and poor prognosis due to its associated morbid conditions.
Key words. Antiepileptic, Electroencephalography, Epilepsy, Lennox-Gastaut syndrome (MeSH).
Introducción
El síndrome de Lennox-Gastaut (SLG) es una encefalopatía epiléptica grave de la niñez que se caracteriza por múltiples tipos de convulsiones (principalmente tónicas, atónicas y ausencias atípicas), electroencefalograma (EEG) con punta onda lenta generalizada (POLG) (<2.5 Hz) en vigilia y paroxismos de ritmos rápidos generalizados (PRRG) durante el sueño, y retraso mental (1-5).
El SLG tiene una incidencia de 2.8 por 10,000 nacidos vivos. Los niños (60%) se afectan más frecuentemente que las niñas. Representa el 3-4% de todas las epilepsias, 10% de las epilepsias refractarias y entre 4% y 8% de las epilepsias en niños menores de 10 años (1-3, 5, 6).
En cuanto a la etiología, el SLG puede ser sintomático o criptogénico. Las formas sintomáticas (70-75%) suelen deberse a trastornos cerebrales, y la mayoría de ellas corresponde a malformaciones o displasias corticales. Las otras causas son la hipoxia-isquemia cerebral pre, peri o postnatal, las anomalías cromosómicas o genéticas como el síndrome de Down, infecciones congénitas o adquiridas, trauma craneoencefálico, hidrocefalia, radioterapia y tumores cerebrales, hemorragia intracraneal, esclerosis tuberosa y errores innatos del metabolismo (2, 3, 5-7).
En las formas criptogénicas (25-30%) no se encuentran factores genéticos predisponentes ni patología cerebral. Las neuroimágenes son normales, al igual que el desarrollo psicomotor hasta el inicio de las crisis. Se puede encontrar historia familiar de epilepsia y puede verse que entre el 10% y el 25% de los pacientes se encuentra el antecedente de síndrome de West (2, 5-7).
Los mecanismos fisiopatológicos subyacentes al desarrollo de esta encefalopatía difusa no se conocen completamente. Debido a que este síndrome cuenta con una gran variedad de causas multifocales o difusas con compromiso cortico-subcortical, actualmente se acepta que el SLG es el resultado de una interacción compleja de estas afecciones que ocurren durante el desarrollo cerebral en un período determinado (2, 3, 5, 8).
El SLG se caracteriza por presentar diversos tipos de crisis; las más características son las tónicas, las ausencias atípicas y las convulsiones atónicas. Se pueden presentar en menor medida otro tipo crisis como las mioclónicas, las tónico-clónicas generalizadas y las focales (2, 6).
En el EEG interictal, la actividad basal es anormal; se encuentra un ritmo alfa lento y fragmentado (70-90%), un exceso de ondas lentas difusas y desorganización electroencefalográfica. El deterioro cognoscitivo es directamente proporcional al grado de lentificación. En el EEG se pueden distinguir los estadios del sueño, aunque no se precisan claramente los husos del sueño, las ondas del vértex y los complejos K (2, 5).
Característicamente se encuentra una actividad de POLG, que consiste en descargas de paroxismos de punta (< 70ms) u onda aguda (70 a 200ms) seguida por una onda lenta sinusoidal de 300 a 500ms. Su frecuencia oscila entre 1Hz y 4Hz (por lo general, entre 1.5Hz y 2.5Hz). Aparecen en forma difusa, aislados o en secuencias variables, con mayor amplitud en las regiones frontales o frontocentrales y son irregulares en cuanto a distribución, amplitud, morfología y frecuencia; por lo general son bilaterales, con cierto grado de asimetría, y de duración variable (3, 5, 9).
El otro patrón distintivo del SLG son los PRRG, que se han asociado a crisis tónicas subclínicas. Se caracterizan por presentar una frecuencia entre 10 y 25Hz con una duración de 1 a 9 segundos; son generalizados, asimétricos y tienen su máxima amplitud en regiones frontales y frontocentrales (3, 5, 9).
En la actualidad no hay un tratamiento eficaz para el SLG. Los diversos tipos de crisis, que son frecuentes y difíciles de cuantificar, y las fluctuaciones de la enfermedad a lo largo del tiempo, hacen que sea difícil evaluar la eficacia de los diversos intentos de tratamiento. Las crisis tónicas son difíciles de tratar mientras que las crisis atónicas, mioclónicas y las ausencias atípicas son más sensibles al tratamiento (2).
Una vez hecho el diagnóstico, el curso irremediable del síndrome es la persistencia de las crisis, la politerapia, la refractariedad y el pobre pronóstico; esto lleva al paciente a un deterioro progresivo cognoscitivo, biológico y psicológico.
Hay divergencias en la información sobre las características clínicas y electroencefalográficas del SLG, en los diferentes artículos publicados de las series evaluadas en el mundo, debido a la falta de uniformidad en los criterios diagnósticos, los grupos poblacionales estudiados y en las variables que se quieren medir (10-22).
No se conocen los rasgos esenciales del comportamiento de este síndrome en nuestro medio, por lo que al caracterizarlo mejor, se pretende que haya un diagnóstico adecuado y temprano. Esto permitirá que se haga un tratamiento, control y seguimiento multidisciplinario del paciente y su familia, que lleven a que disminuya la carga de la enfermedad y sus complicaciones.
Consecuentemente, el objetivo del estudio es describir las características clínicas y electroencefalográficas en una muestra de pacientes con síndrome de Lennox-Gastaut diagnosticados en el programa de epilepsia de la Universidad de Antioquia en Medellín entre 2007 y 2012.
Materiales y Métodos
Se trata de un estudio observacional, descriptivo y retrospectivo. La población de estudio incluyó todos los registros de pacientes con diagnóstico de síndrome de Lennox-Gastaut contenidos en la base de datos del programa de epilepsia de la Universidad de Antioquia y que fueron evaluados por videomonitoreo electroencefalográfico. La historia clínica de cada paciente fue diligenciada por uno de los coautores (RASM). La muestra está constituida por 18 registros de pacientes evaluados en el programa de epilepsia de la Universidad de Antioquia y que están consignados en la base de datos. En cuanto al muestreo, la inclusión de los registros se hizo de manera no aleatoria y por conveniencia de aquellos registros que cumplieron los criterios de inclusión.
Criterios de inclusión: pacientes registrados de cualquier edad que tuvieran el diagnóstico electroclínico de síndrome de Lennox-Gastaut, de acuerdo con los siguientes criterios: 1) múltiples tipos de convulsiones principalmente tónicas, atónicas y ausencias atípicas, 2) electroencefalograma (EEG) con punta onda lenta generalizada (POLG) (<2.5 Hz) en vigilia y paroxismos de ritmos rápidos generalizados (PRRG) durante el sueño, y 3) retraso mental y/o trastornos comportamentales. Además de satisfacer los mencionados criterios, fue necesario contar con la información sociodemográfica, clínica y las telemetrías para revisión.
Criterios de exclusión: no disponibilidad de información suficiente o completa para el análisis en los registros.
Procedimientos: se revisó la base de datos del laboratorio de neurofisiología, electroencefalografía y telemetría CEC-LAB de la Clínica León XIII (IPS Universidad de Antioquia). En los registros con impresión diagnóstica de síndrome de Lennox-Gastaut, se verificó que cumplieran con los criterios diagnósticos de inclusión establecidos para el presente estudio. Para los que cumplieron estos criterios, se revisó la información disponible en la historia clínica y se analizaron las telemetrías. La información obtenida se consignó en un formulario que incluía las variables seleccionadas para este fin en la investigación. Los registros que estaban incompletos fueron excluidos. Los datos contenidos en los formularios se digitaron en una base de datos en Excel que después se exportó al programa estadístico SPSS versión 15.0 para su análisis.
Plan de análisis: se realizó un análisis descriptivo de los datos consignados en el formulario. Para las variables de tipo cualitativo, se hizo un análisis de frecuencias con sus respectivas proporciones, y para las cuantitativas se calcularon los promedios con su respectiva desviación estándar. En caso de que la distribución de la variable no fue normal, se calcularon la moda, la mediana y los rangos.
Aspectos éticos: este trabajo fue aprobado por el Comité de Ética de la Universidad de Antioquia y no trajo beneficios personales o económicos a los autores.
Resultados
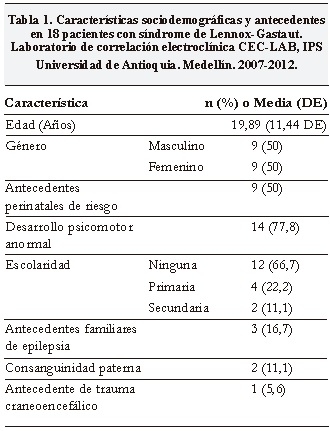
Se revisaron 18 videotelemetrías con diagnóstico clínico y electroencefalográfico de SLG. La edad promedio de los pacientes fue de 19.89 ±11.44 años con edades comprendidas entre los 3 y 44 años y con igual proporción entre sexos.
La mitad de los pacientes presentaba antecedentes perinatales de riesgo durante el embarazo y/o el parto dados en su mayoría por amenazas de aborto y de parto pretérmino, prematurez o sufrimiento fetal agudo.
Solo 6 pacientes (33,3%) tuvieron algún grado de escolaridad y la mayoría de ellos (77,8%) tuvo un desarrollo psicomotor anormal (Tabla 1).
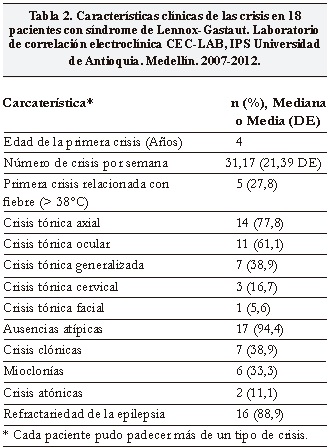
La mediana de la primera crisis fue de 4 años con rango de un mes a quince años y el número promedio de crisis por semana fue de 31.17±21.39
Las crisis más frecuentes fueron las ausencias atípicas en 17 pacientes (94,4%), tónica axial en 14 (77,8%) y tónica ocular en 11 (61,1%). Un 27,8% de los casos tenía antecedente de convulsiones febriles. Se observó refractariedad de la epilepsia, definida en nuestro estudio como la ausencia de control de crisis con tres fármacos a dosis plenas, en 16 pacientes (88,9%) (Tabla 2).
En lo que se refiere al tratamiento administrado en algún momento de la evolución, los medicamentos más utilizados fueron el ácido valproico en 17 pacientes (94,4%), el fenobarbital en 12 (66,7%) y la carbamazepina y el levetiracetam en 11 (61,1%). Adicionalmente y de manera preocupante se encontró tratamiento para el SLG con medicamentos contraindicados para dicha entidad en más del el 50% de de los casos (Tabla 3).

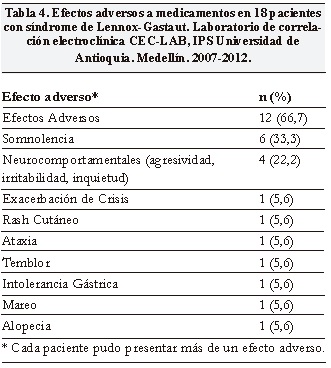
Se encontraron efectos adversos a medicamentos anticonvulsivantes en 12 pacientes (66,7%); de estos, los más frecuentes fueron: somnolencia en 6 (33,3%) y efectos neurocomportamentales (agresividad, irritabilidad e inquietud) en 4 (22,2%) (Tabla 4).
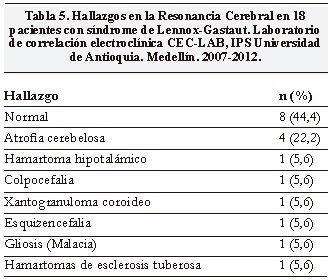
Se realizaron resonancias cerebrales a todos los pacientes, siendo normal en 8 (44,4%) y con atrofia cerebelosa en 4 (22,2%). Otros hallazgos imagenológicos pueden ser verse en la Tabla 5.
Se realizó valoración neuropsicológica a dos pacientes (11,1%), y se encontró en ellos retardo mental leve. En los restantes 16 pacientes se diagnosticó retardo mental por historia clínica y examen neurológico.
Se encontraron seis casos criptogénicos (33,3%) definidos por la ausencia de factores de riesgo pre, peri o postnatales IRM cerebral normal. En los doce casos sintomáticos (66,7%) se encontraron antecedentes de meningitis, prematurez, sufrimiento fetal agudo o hallazgos anormales en la IRM cerebral (Tabla 5).
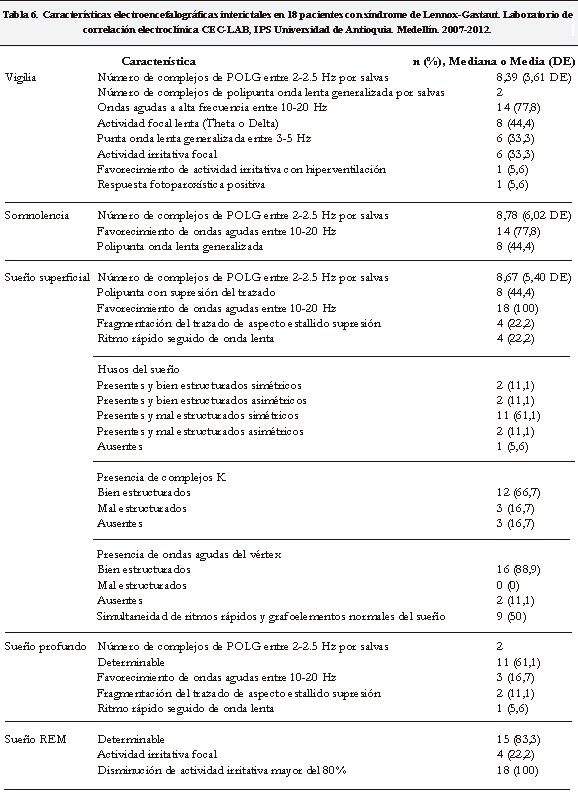
En cuanto a los hallazgos electroencefalográficos interictales, en vigilia se encontró actividad de punta onda lenta generalizada de 2 a 2.5Hz en todos los pacientes y PRRG en 14 de ellos (77,8%). Esta actividad no fue modificada por las maniobras de activación, como la hiperventilación o la fotoestimulación, pero aumentó y se vio favorecida durante el sueño no REM, especialmente durante el sueño superficial. En este estado se observaron la mayoría de anormalidades. En el 30% de los registros electroencefalograficos se observó actividad irritativa focal, además de la actividad generalizada.
Durante el sueño superficial, los husos del sueño, se presentaron de manera simétrica y bien estructurada, solo en el 11% de los pacientes. El resto de los registros tenía anormalidades en este grafoelemento por asimetría (11%) o mala estructuración de los mismo (61,1%). Los complejos K y las ondas agudas del vértex, se encontraron bien estructuradas en 12 (66,7%) y 16 pacientes (88,9%) respectivamente.
El número promedio de complejos de POLG en los registros de sueño y vigilia fueron 8.57±5.4 y 8.39±3.61 respectivamente.
En el trazado del ritmo de fondo se encontró fragmentación con aspecto de estallido supresión, dado por la presencia de actividad de alto voltaje seguida de supresión del trazado mayor de 5mV de los registros, al igual que un ritmo caracterizado por actividad rápida seguida de onda lenta en la misma proporción de casos.
El sueño REM estuvo caracterizado por una disminución notable en su duración (en promedio menor de 30 minutos) y por una disminución de la actividad irritativa mayor del 80% en todos los trazados revisados. Adicionalmente, en el 22% de los registros se encontró persistencia de actividad irritativa focal en esta etapa del sueño (Tabla 6).
Los hallazgos electroencefalográficos ictales se enmarcaron por la presencia de crisis de ausencias atípicas en la mayoría de pacientes, 14 (77,8%). Por su parte, la presencia de microcrisis reclutantes se dio en el 100% de los pacientes durante el sueño, pero hasta en el 66,7% de los casos se dio en vigilia. No se desmostró favorecimiento de crisis tónicas o mioclónicas con la fotoestimulación; solo en un regsitro se apreció lentificación del trazado. Durante el sueño REM no se vieron eventos motores tónicos o tónico-clónicos (Tabla 7).

Discusión
En la literatura médica se encuentran varios estudios que describen las características sociodemográficas, clínicas y electroencefalográficas del SLG (10-22). En cuanto a las características sociodemográficas, las publicaciones realizadas previamente muestran un claro predominio del género masculino (10-22); solamente Ferlazzo et al. (18) en una investigación sobre pronóstico del SLG en 27 pacientes adultos, encontraron un leve predominio de mujeres (15 mujeres frente a 12 hombres). En nuestro estudio se obtuvo la misma proporción de hombres y mujeres (9 cada uno). Al igual que el estudio de Ferlazzo et al., posiblemente el pequeño tamaño de la muestra no fue suficiente para corroborar los datos epidemiológicos previos; a saber, que los hombres se afectan más frecuentemente que las mujeres (1-3, 5, 6).
Los factores perinatales de riesgo se relacionan fuertemente con la ocurrencia de hipoxia-isquemia cerebral, que es una de las causas importante de SLG sintomático (2, 3, 5-7). El hallazgo de antecedentes perinatales de riesgo en el 50% de los pacientes, es consistente con lo reportado por Oguni et al. (11), quienes encontraron antecedentes pre y perinatales de riesgo en 44 de 72 pacientes (61,1%), y por lo descrito por Heiskala (14) respecto de los factores de riesgo pre y perinatales en el 49% y 41% de sus pacientes, respectivamente.
En el mal pronóstico cognoscitivo de los pacientes con SLG, no solo interviene la presencia progresiva de retardo mental, sino también el antecedente de retraso del desarrollo al inicio del síndrome (16). Heiskala (14) encontró el antecedente de retraso en el desarrollo en 22 de 73 pacientes (30,1%) con SLG; este hallazgo se correlacionó de forma significativa con una epilepsia más severa. Los estudios conducidos por Goldsmith et al. (16) y Rodríguez et al. (20) también encontraron este antecedente en el 8,4% y 33,3% de sus pacientes respectivamente. Nuestro hallazgo de retardo del desarrollo del 77,8% solo es comparable con el estudio de Herranz et al. (19) donde el retraso del desarrollo representó el 79% en 331 pacientes. Este mayor porcentaje de pacientes con desarrollo psicomotor anormal en nuestro estudio puede deberse a que incluimos en la categoría de retraso del desarrollo tanto a pacientes con trastornos motrices, como a pacientes con trastornos del aprendizaje y del lenguaje.
En el SLG es posible encontrar historia familiar de epilepsia entre el 3% y el 30% de los casos (2, 5-7), antecedente que se puede asociar con una epilepsia más favorable (14). En la presente serie se encontraron antecedentes familiares de epilepsia en el 16,7% de los pacientes, porcentaje que coincide con los datos epidemiológicos de la literatura y con los resultados de los estudios de Heiskala (14) (29%), Goldsmith et al. (16) (13,1%) y Hoffmann-Riem et al. (17) (30,6%). Sin embargo, Rodríguez et al. (20) no encontraron antecedentes de epilepsia en la familia de 12 pacientes con SLG posiblemente debido al tamaño de la muestra.
La edad de inicio del SLG oscila entre los 1 y 8 años, con un pico entre los 3 y 5 años (1-3, 5, 6). Se ha encontrado además que un inicio temprano de las crisis en el SLG se correlaciona con la severidad de la epilepsia, un alto riesgo de convulsiones intratables y peor pronóstico neurológico (13, 14). La mediana de 4 años como edad de la primera crisis es consistente con lo reportado en estudios previos (11, 12, 15-20).
El SLG está clasificado como una de las formas más graves de epilepsia dada la alta frecuencia y severidad de las crisis; las más frecuentes en su orden son las tónicas (80-100%), ausencias atípicas (17-60%) y atónicas (25-56%) (2, 6). Las crisis más frecuentes en los pacientes de este estudio fueron las tónicas, principalmente las axiales (77,8%), y las ausencias atípicas (94,4%); estos datos son consistentes con lo reportado en estudios previos (11, 16-20). Llama la atención que las crisis atónicas solo fueron reportadas en dos pacientes (11,1%); esto posiblemente corresponda a que las crisis tónicas del cuello que se reportaron se debieron a crisis atónicas que comprometieron solo el tono muscular cefálico o que las caídas, generalmente con traumas, presentadas en estos pacientes fueran referidas por los familiares más como crisis tónicas. Esto último puede deberse a que, al igual que estas, las crisis atónicas causan una gran cantidad de desplomes al suelo. Herranz et al. (19) encontraron en 302 pacientes con SLG una frecuencia de crisis diaria en 166 (55%), semanal en 70 (23%) y mensual en 66 (22%). Al considerar todos los casos conjuntamente, la media de crisis fue de 4 ± 6,2 crisis diarias (rango: 0-50), lo que supone una media semanal de 27,9 ± 43,7 (rango: 0-350). Nuestro hallazgo de una media de 31,17 (21,19 DE) crisis por semana es comparable con estos resultados y demuestra la alta frecuencia de crisis en estos pacientes.
El antecedente de convulsiones febriles en un 27% de casos es un hallazgo inesperado y de significado incierto.
Esta gran diversidad de las crisis, unida a su alta frecuencia, lleva a otra característica del SLG, a saber, su refractariedad. Esto, porque se utilizan varios medicamentos de amplio espectro a altas dosis que a menudo incrementa los efectos adversos. La refractariedad del SLG fue encontrada en el 88,9% de los pacientes.
La politerapia es característica del SLG y se ha reportado en estudios previos (14, 15, 17, 19, 20). El fármaco más utilizado en algún momento de la evolución en estos pacientes fue el ácido valproico en un 94,4% de los casos, lo que también es visto en estudios anteriores (14, 18-20). Sin embargo, en estos estudios también se observa después del ácido valproico, alta frecuencia en el uso de otros fármacos como la lamotrigina, el topiramato y el clobazam, que en esta serie fueron reemplazos en orden de frecuencia con fármacos como el fenobarbital (66,7%), la carbamazepina (61,1%) y la fenitoína (50%). Estos últimos medicamentos son administrados con mayor facilidad por el sistema de salud. Se encontró que en la mitad de los casos los pacientes recibieron, en algún momento de su evolución, medicamentos que están contraindicados para este síndrome por estar asociado su empleo con la exacerbación de las crisis o el empeoramiento de la encefalopatía. De otro lado, llama la atención que a pesar de una alta proporción de refractariedad no se ofrecieron otras opciones terapéuticas como dieta cetogénica o cirugía.
El predominio en nuestro estudio de los pacientes con SLG sintomático frente a los criptogénicos (66,7 vs 33,3%) es similar a lo reportado en la literatura (11, 13, 15-17, 19). Los hallazgos anormales en la neuroimágenes explican una buena parte de los casos sintomáticos e incluso se relacionan con una epilepsia más severa como lo reporta en su estudio Heiskala (14). En los casos sintomáticos encontramos anomalías en la IRM cerebral como esquizencefalia y hamartomas de esclerosis tuberosa, y antecedentes perinatales de riesgo y de infección del SNC que también se han informado previamente (13, 15-17, 21). Esto pone de manifiesto la necesidad de un diagnóstico oportuno y de un tratamiento precoz de las neuroinfecciones en la edad pediátrica, así como de un adecuado control prenatal y atención del trabajo de parto para minimizar la ocurrencia de estas causas del SLG. Otro aspecto importante en la etiología, es que estudios previos han reportado antecedente de síndrome de West en porcentajes que van del 18,5% al 40% (11, 12, 14-18, 20, 21). En este estudio solo un paciente tuvo este antecedente (5,5%), posiblemente debido a que la cantidad de pacientes no fue suficiente para representar a toda la población. Goldsmith et al. (16) aluden también este sesgo en su estudio al encontrar un porcentaje bajo de pacientes con síndrome de West previo (13,1%).
Otra característica importante y criterio diagnóstico del SLG es el retardo mental, especialmente más marcado en pacientes sintomáticos o con antecedente de síndrome de West (10, 11, 15). Estudios previos coinciden en mostrar altos porcentajes de retardo mental en estos pacientes con cifras que van del 91% al 100% (10, 13, 16-20). Este factor de mal pronóstico también se encontró en todos los pacientes evaluados en nuestro estudio, aunque en solo dos de ellos se constató por valoración neuropsicológica.
Este déficit cognoscitivo altera todos los aspectos de la vida de la persona con SLG, especialmente el académico. En el estudio se encontró que el 66,7% de los pacientes no tuvo ningún grado de escolaridad y los restantes no culminaron satisfactoriamente su primaria y secundaria o presentaron repetición de años y fracaso escolar. A este respecto Herranz et al. (19) describen en su estudio que el 62% de los pacientes estaba escolarizado en centros de educación especial y el 11% en centros ordinarios de integración. Los trastornos conductuales y psiquiátricos que se suman al retardo mental afectan también al entorno familiar. Ferlazzo et al. (18) en 27 pacientes con SLG encontraron que, aunque 12 vivían con su familia, 15 tuvieron que ser institucionalizados.
En todos los pacientes del estudio se encontraron los hallazgos electroencefalográficos característicos del SLG como son la actividad de POLG (<2.5Hz) en vigilia y PRRG durante el sueño, principalmente favorecidos durante el sueño superficial. Dependiendo de la definición y de los criterios de inclusión utilizados, la mayoría de los pacientes de estudios previos también presentaron en algún momento de la evolución la actividad de POLG o los PRRG propios del SLG (11, 14, 17- 20). Esto pone de manifiesto la importancia de un adecuado seguimiento de estos pacientes, no solo clínico sino también electroencefalográfico, ya que algunos pacientes en las etapas iniciales pueden no presentar los rasgos característicos del SLG, llevando a postergar un diagnóstico y tratamiento precoz (1).
Los hallazgos electroencefalográficos durante el sueño constituyen el centro de la discusión. La arquitectura del sueño está alterada por la asimetría o mala estructuración de los husos del sueño y disminución del tiempo de sueño REM. Lo anterior se encuentra relacionado con discapacidad cognoscitiva y un pobre pronóstico funcional, como se ha descrito en otras series y en casos de otras encefalopatías epilépticas (27).
Se comprobó la presencia de descargas de POL que superaban frecuencias de 2.5Hz, sin que implicara cambios clínicos o sobre otras variables electroencefalográficas.
Se demostró la existencia de ritmos rápidos seguidos de otros elementos del sueño, pero se desconocen sus implicaciones pronósticas. De forma similar a lo contenido en otras publicaciones se evidenció actividad ictal e interictal de tipo focal.
Un hallazgo que se destaca es la observación de ritmos rápidos en vigilia, pues esto contrasta con lo descrito clásicamente como una característica típica en sueño.
En conclusión, el SLG es una de las encefalopatías epilépticas más severas de inicio en la niñez caracterizada por la presencia de varios tipos de crisis, principalmente las tónicas y las ausencias atípicas. Estas crisis son severas, frecuentes y refractarias al tratamiento, aunado a un compromiso de la función cortical con desestructuración severa de los ritmos fisiológicos de todas la etapas del sueño NREM con disminución en tiempo del sueño REM más la lentificación difusa moderada en vigilia. Lo anterior conlleva grandes costos sociales y económicos para el paciente y su familia debido a su alta morbilidad y asociación con retardo mental y trastornos psiquiátricos y conductuales. Nuevos estudios de mayor complejidad son requeridos para profundizar en las causas y el pronóstico del SLG y sus condiciones mórbidas asociadas.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Referencias
1. ARZIMANOGLOU A, FRENCH J, BLUME WT, CROSS JH, ERNST JP, FEUCHT M, ET AL. Lennox-Gastaut syndrome: a consensus approach on diagnosis, assessment, management, and trial methodology. Lancet Neurol 2009; 8(1): 82-93. [ Links ]
2. PANAYIOTOPOULOS CP. Epileptic encephalopathies in infancy and early childhood. In: Panayiotopoulos CP. A clinical guide to epileptic syndromes and their treatment. 2ª ed. Londres: Springer, 2010: 275-326. [ Links ]
3. MARKAND ON. Lennox-Gastaut syndrome (childhood epileptic encephalopathy). J Clin Neurophysiol 2003; 20(6): 426-41. [ Links ]
4. CRUMRINE PK. Lennox-Gastaut syndrome. J Child Neurol 2002; 17(Suppl.): S70-5. [ Links ]
5. BERMÚDEZ MALDONADO L, MORENO AVELLÁN AJ. Síndrome de Lennox-Gastaut. El Residente 2009; 4(2): 56-66. [ Links ]
6. ARCHILA R, PAPAZIAN O. Síndrome de Lennox-Gastaut. Rev Neurol 1999; 29(4): 346-9. [ Links ]
7. CAMFIELD PR. Definition and natural history of Lennox-Gastaut syndrome. Epilepsia 2011; 52(Suppl. 5): 3-9. [ Links ]
8. BLUME WT. Pathogenesis of Lennox-Gastaut syndrome: considerations and hypotheses. Epileptic Disord 2001; 3(4): 183-96. [ Links ]
9. HRACHOVY RA, FROST JD JR. The EEG in selected generalized seizures. J Clin Neurophysiol 2006; 23(4): 312-32. [ Links ]
10. OHTSUKA Y, AMANO R, MIZUKAWA M, OHTAHARA S. Long-term prognosis of the Lennox-Gastaut syndrome. Jpn J Psychiatry Neurol 1990; 44(2): 257-64. [ Links ]
11. OGUNI H, HAYASHI K, OSAWA M. Long-term prognosis of Lennox-Gastaut syndrome. Epilepsia 1996; 37(Suppl. 3): 44-7. [ Links ]
12. YAGI K. Evolution of Lennox-Gastaut syndrome: A long-term longitudinal study. Epilepsia 1996; 37(Suppl. 3): 48-51. [ Links ]
13. TREVATHAN E, MURPHY CC, YEARGIN-ALLSOPP M. Prevalence and descriptive epidemiology of Lennox-Gastaut syndrome among Atlanta children. Epilepsia 1997; 38(12): 1283-8. [ Links ]
14. HEISKALA H. Community-based study of Lennox-Gastaut syndrome. Epilepsia 1997; 38(5): 526-31. [ Links ]
15. RANTALA H, PUTKONEN T. Occurrence, outcome and prognostic factors of infantile spasms and Lennox-Gastaut syndrome. Epilepsia 1999; 40(3): 286-9. [ Links ]
16. GOLDSMITH IL, ZUPANC ML, BUCHHALTER JR. Long-term seizure outcome in 74 patients with Lennox-Gastaut syndrome: effects of incorporating MRI head imaging in defining the cryptogenic subgroup. Epilepsia 2000; 41(4): 395-9. [ Links ]
17. HOFFMANN-RIEM M, DIENER W, BENNINGER C, RATING D, UNNEBRINK K, STEPHANI U, ET AL. Nonconvulsive status epilepticus-a possible cause of mental retardation in patients with Lennox-Gastaut syndrome. Neuropediatrics 2000; 31(4): 169-74. [ Links ]
18. FERLAZZO E, NIKANOROVA M, ITALIANO D, BUREAU M, DRAVET C, CALARESE T, ET AL. Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Res 2010; 89(2-3): 271-7. [ Links ]
19. HERRANZ JL, CASAS-FERNÁNDEZ C, CAMPISTOL J, CAMPOS-CASTELLÓ J, RUFO-CAMPOS M, TORRES-FALCÓN A, ET AL. Síndrome de Lennox-Gastaut en España: estudio epidemiológico retrospectivo y descriptivo. Rev Neurol 2010; 50(12): 711-7. [ Links ]
20. RODRÍGUEZ-RODRÍGUEZ S, SALAS-PUIG J, ÁLVAREZ-CARRILES JC, TEMPRANO-FERNÁNDEZ T, ANTÓN-GONZÁLEZ C, GARCÍA-MARTÍNEZ A. Evolución del síndrome de Lennox-Gastaut en la edad adulta. Rev Neurol 2011; 52(5): 257-63. [ Links ]
21. VALDIVIA ÁLVAREZ CI, MARRERO MARTÍNEZ P. Caracterización etiológica del síndrome de Lennox-Gastaut sintomático. Revista Cubana de Pediatría 2012; 84(1): 22-32. [ Links ]
22. KERR M, KLUGER G, PHILIP S. Evolution and management of Lennox-Gastaut syndrome through adolescence and into adulthood: are seizures always the primary issue? Epileptic Disord 2011; 13(Suppl. 1): S15-26. [ Links ]
