










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.1 Bogotá Jan./Mar. 2015
https://doi.org/10.22379/2422402213
Revisión
Fisiopatología de la migraña: Teoría vascular, ¿Cierta o no?
Pathophysiology of migraine: vascular theory, true or not?
David Benavides (1), Laura Cristina Rodríguez (1), Jorge Restrepo (2), Daniela Vargas B. (1)
(1) Estudiantes de Medicina.
(2) Profesor de Neurología. Universidad de La Sabana. Chía, Colombia.
Recibido: 17/03/14. Aceptado: 6/01/15.
Correspondencia: Jorge Restrepo: jare7000@yahoo.com
Resumen
La migraña es una enfermedad de alta prevalencia, incapacitante y en algunas ocasiones de difícil manejo. Desde hace décadas se han planteado múltiples teorías para explicar su curso, su componente genético y la asociación a distintos factores de riesgo. Actualmente se desconoce una fisiopatología única y exacta que implique los eventos, y se ha encontrado fuerte evidencia que muestra que la teoría más antigua y con mayor sustento, a saber la teoría vascular, es en realidad incorrecta, pues no explica la totalidad de los eventos. Sin embargo, se han probado distintos mecanismos que, en conjunto, permiten comprender las alteraciones presentes. Entre estas se cuentan cambios estructurales, implicación de neuropéptidos, sensibilización, e inflamación neurogénica.
Palabra clave: Migraña, Péptido relacionado con el gen de la calcitonina (PRGC), Sistema trigémino, sensibilización, glutamato, inflamación neurogénica, teoría vascular (DeCS).
Summary
Migraine is a highly prevalent disease; it is disabling, and sometimes difficult to manage. For decades, many theories have been proposed to explain its course, its association with a genetic component and with different risk factors. There is currently no single and exact pathophysiology that accounts for all events, and strong evidence has been showing that the oldest theory believed to be mostly true, i.e. the vascular theory, is actually incorrect, because it does not explain the totality of the events. However, various mechanisms have been proven to exist, which together, provide insight into alterations, such as structural changes involving neuropeptides, sensitization, and neurogenic inflammation.
Key words. Migraine, Calcitonin Gene-Related Peptide, Serotonin (MeSH), Trigeminal System, sensitization, glutamate, neurogenic inflammation, vascular theory (MeSH).
Introducción
La fisiopatología de la migraña ha sido estudiada por décadas y, no obstante, aún sigue siendo tema de discusión y de controversia cuál sea el mecanismo exacto que pueda explicar en su totalidad los eventos que generan el dolor, el aura y la cascada de eventos subsiguientes. Se han encontrado diversos factores de riesgo y agravantes en ataques de migraña como los siguientes: ser mujer, tener un bajo nivel socioeconómico, haber sufrido de trauma craneoencefálico, tener síndrome de apnea-hipopnea obstructivo del sueño, consumir cafeína y abusar de analgésicos. También se sabe que la obesidad con un IMC >30 aumenta cinco veces el riesgo de padecer migraña (1). Además de los factores ya mencionados, se ha encontrado un riesgo aumentado en pacientes con las siguientes condiciones: foramen oval permeable, isquemias cerebrales que potencialmente causen microembolias a repetición e hipoperfusión transitoria, tales como el síndrome de CADASIL, Síndrome de Sjogren, trombocitosis, policitemia vera, malformaciones arteriovenosas, AIT (accidente isquémico transitorio) y posterior a un ataque cerebrovascular tanto hemorrágico como isquémico. Puede notarse que fisiopatológicamente estos factores de riesgo conducirían a una hipoperfusión transitoria con alteración de los vasos cerebrales, llevando a una vasoconstricción primaria con posterior vasodilatación, de forma que se desarrolle la ya conocida depresión cortical propagada (2). Sumado a esto, recientemente se ha replanteado y discutido nueva información respecto de aspectos conocidos de la fisiopatología como la inflamación neurogénica, la sensibilización, los neuropéptidos, el péptido relacionado con la calcitonina, los cambios estructurales, y el glutamato (3).
Este artículo pretende hacer una revisión de la literatura para encontrar información suficiente que apoye factores a favor o en contra respecto de la teoría vascular en la migraña. Así mismo, se propone plantear otras variantes implicadas en su fisiopatología, tal vez menos reconocidas, que pueden ser de igual o mayor importancia que el sistema clásico vascular que se acepta en la actualidad para la explicación.
TEORÍA TRIGÉMINO-VASCULAR
Desde hace un tiempo se conoce la relación existente entre la migraña y la cefalea en general con el sistema trigémino vascular; se conocen bien la anatomía y la fisiología del dolor cráneo-facial, las vías nerviosas y los núcleos implicados (Figura 1). No obstante, aún es incierta la causa o el evento desencadenante que dispara la cascada de reacciones nerviosas que finalmente generan el dolor de cabeza (4).
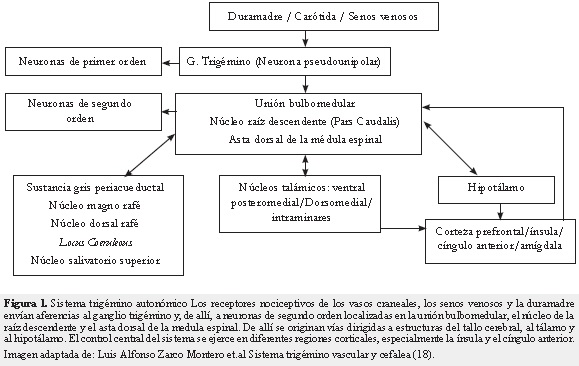
La migraña se reconoce por una serie de eventos que desencadenan una respuesta nerviosa trigeminal, sobre una ya estudiada predisposición genética. El componente hereditario de la migraña es evidente en la práctica clínica donde se encuentran patrones y tipos específicos de cuadros migrañosos (5). Entre estos se pueden caracterizar las diferentes variantes de la migraña familiar hemipléjica como la tipo 1 (MHF 1), que posee una mutación del cromosoma 19p13 y se caracteriza por una disfunción de los canales de calcio voltaje dependientes que son las estructuras principales en el flujo de calcio neuronal, con lo que establece un papel crítico en la señalización neuronal (6).
Los canales Cav2 tipo P/Q (alto voltaje), se encuentran en la zona activa de las terminales presinápticas excitatorias y se encargan de la liberación rápida de neurotransmisores. El gen CACNA1A codifica la subunidad α1 del canal de calcio dependiente de voltaje tipo CaV2.1 (P/Q), y las mutaciones en esta subunidad causan la entrada de calcio a la neurona y una respuesta a pequeñas despolarizaciones, lo cual genera una liberación excesiva de glutamato (26).
La migraña familiar hemipléjica tipo 2 (MHF 2) se manifiesta por una mutación en el gen ATP1A2 que codifica para la subunidad α2 de la bomba Na+/K+ ATPasa, que se encuentra localizada en el cromosoma 1q23. Dichas mutaciones generan cambios en los dominios transmembrana de la bomba, las cuales determinan la ausencia en su función; esto genera una elevación de la concentración de K+ extracelular y por ende hiperpolarización de la célula (21).
La migraña familiar hemipléjica tipo 3 (MHF3) está asociada al gen SCN1A, localizado en el cromosoma 2q24 que codifica para la subunidad α1 de los canales de sodio voltaje dependiente tipo NaV1.1. Las mutaciones en estos genes conllevan la pérdida de la actividad del canal de sodio NaV1.1, inicialmente en neuronas inhibitorias alterando así su funcionalidad. No obstante esta pérdida, se aumenta la actividad de neuronas excitatorias y de esta manera se elevan las concentraciones de glutamato y potasio en la hendidura sináptica. Este hecho se ha relacionado finalmente como un facilitador de la generación de la onda de depresión cortical (6).
La teoría vascular de la migraña plantea que existe una serie de eventos que se desencadenan por un factor que produce una microembolia o isquemia focal transitoria dentro del sistema nervioso central (7, 8). Esta alteración de los vasos cerebrales dispara una onda lenta de propagación de despolarización neuronal y glial en la corteza, cerebelo, ganglios basales, hipocampo y tálamo llamada propagación de depresión cortical (PDC) descrita inicialmente por Leao. Este fenómeno fue relacionado con la extensión de una depresión funcional neuronal que se inicia en la región occipital y progresa hacia la región anterior; la hipoperfusión cortical y meningea también está relacionada con la consiguiente disminución del metabolismo cortical (15), la cual posteriormente activará las neuronas sensitivas del sistema trigémino que provocarán el dolor.
Por otro lado, la fisiopatología del dolor y la propagación de depresión cortical son causadas por cambios que se presentan en el tono vascular donde hay una liberación neuronal y endotelial de neurotransmisores que estimulan las terminales periféricas del nervio trigémino. Esta se manifiesta con la dilatación de las arterias durales y piales (11) y la liberación de otras sustancias que ayudan a propagar la respuesta como el péptido relacionado con el gen de la calcitonina (PRGC) y el polipéptido intestinal vasoactivo (VIP). Estos promueven la inflamación y posteriormente se presenta vasodilatación, extravasación de proteínas, cambios celulares y del endotelio, agregación plaquetaria, liberación de serotonina, sustancia P, CGRP y la degranulación de células mastocitarias. Estas últimas pueden activar sustancias que estimulan las células nerviosas nociceptivas que transmiten información a lo largo de las fibras trigéminovasculares y así propagan la respuesta inflamatoria y transmiten la información hacia el núcleo caudalis del trigémino y los centros sensitivos cerebrales superiores (20) (Figura 2).
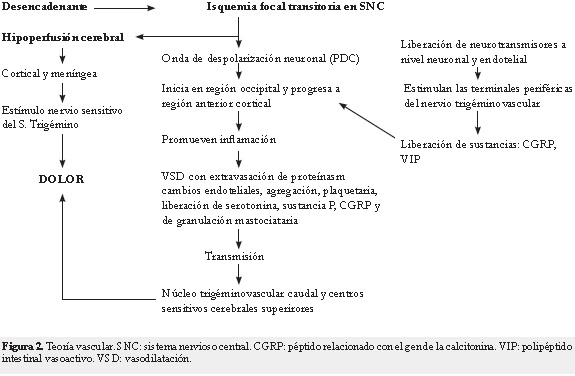
Se han encontrado también diversos factores como el glutamato, otras hormonas y factores genéticos que puedan activar la PDC, y el episodio de cefalea. La isquemia asociada a los ataques de migraña no muestra ningún signo de necrosis o daño histológico en estudios que se han hecho con ratones (12).
La migraña ha sido relacionada con la activación de una variedad de mecanismos moleculares y genes autorreguladores como los que codifican para la ciclooxigenasa 2 (COX2), el factor de necrosis tumoral alfa (TNF), la interleukina1beta, galanina, y las metaloproteinasas. La activación de estas metaloproteinasas conduce a fugas en la barrera hematoencefálica, permitiendo el paso de sustancias como potasio, óxido nítrico, la adenosina, y otros productos liberados por dichos genes que alcanzan y sensibilizan las terminaciones nerviosas perivasculares trigémino aferentes de la duramadre.
El aumento de la actividad de las metaloproteinasas-2 (MMP2) de la matriz ha sido demostrada en pacientes con migraña. Los pacientes que tienen migraña sin aura parecen tener una mayor proporción de metaloproteinasa-9 (MMP9) en la matriz e inhibidores de la metaloproteinasa-1 (TIMP1), en contraste con una relación inferior de MMP9 / TIMP1, en pacientes que tienen migraña con aura.
Además de la asociación que presenta esta teoría con los diversos factores de riesgo ya presentados, existen otros factores que han contribuido a que se haya propuesto como verdadera. Entre estos se encuentra la reciente observación de una presencia significativa de una mayor cantidad de agregados plaquetarios y leucocitarios en el plasma en los pacientes con un diagnóstico previo de migraña; también se halló un incremento sérico del factor de Von Willebrand (9,10).
Sin embargo, a pesar de verse relacionada la alteración del flujo cerebral y endotelial con esta patología, se ha encontrado que en enfermedades en las que debería existir un mayor riesgo de migraña debido a su fisiopatología y al aumento de estados procoagulantes y disfunción endotelial, como el síndrome anti fosfolípidos, no aumenta el riesgo para desencadenar migraña con o sin aura (12).
Algunos autores han observado que a pesar de la relación teórica que existe entre los factores de riesgo vasculares con la migraña, sólo se han encontrado lesiones parecidas a infarto e isquemias transitorias en el tipo de migraña que tiene aura, y no en la migraña sin aura. Es más, los estudios más novedosos con hechos concisos han demostrado claramente que hay una relación muy escasa entre estos dos sucesos. Se han hecho ensayos clínicos con sustancias vasodilatadoras craneales que no muestran que aumenten los episodios de migraña o que los produzcan (12, 13). De igual manera, en un estudio reciente en pacientes a los que se había inducido migraña con nitroglicerina, se sostuvo la la hipótesis de que la migraña respondía a una vasodilatación, pero con angioresonancia de vasos extra e intracraneales, no se halló ninguna alteración en estos (13, 14). Recientemente se ha afirmado que en ataques agudos de dolor unilateral migrañoso sin aura, no hay definitivamente diferencia de tamaño y diámetro en arterias tanto extracraneales como intracraneales (15).
Estos resultados curiosamente entran en contradicción con la respuesta muy eficiente de los pacientes a medicamentos que fueron creados como vasoconstrictores cerebrales, tales como los triptanes y los inhibidores de serotonina principalmente (16). Esto hace pensar que tales medicamentos no disminuyen el dolor por el mecanismo pensado inicialmente sino, más bien, por la transmisión de neurotransmisores del sistema trigémino; para afirmar tal tesis, no obstante, se requieren estudios más a fondo. Es por eso que actualmente sólo se considera que la teoría vascular, con sus determinantes como la microembolia y la isquemia transitoria, solo se comporta como un factor asociado a la migraña con aura. Tampoco se podría afirmar que la isquemia sea la causa del dolor y del aura, pues esto no ha sido comprobado con un estudio; en cambio, habría que afirmar que están relacionados y que esta es un factor de riesgo más para desarrollar posteriormente la patología (17).
De acuerdo con lo planteado anteriormente, se deben tener en cuenta otros mecanismos de acción haciendo énfasis en los neurotransmisores, en el gen relacionado con el péptido de la calcitonina (PRGC) y en el glutamato, los cuales actúan sin efectos dilatadores. Es necesario, además, contar con el efecto de la sensibilización neuronal con lo que se puede proponer que se dejen a un lado solo los efectos vasculares antes pensados.
En la Tabla 1 se enuncia las diferencias entre la teoría vascular y neurovascular de la migraña.

NEUROTRANSMISORES:
Conforme se desarrolla la migraña en un paciente, encontramos diferentes sustancias mediadoras provenientes de ramas simpáticas, parasimpáticas y sensitivas que inervan estructuras craneanas, como los vasos y las leptomeninges. Las fibras simpáticas que provienen del ganglio cervical superior liberan neuropeptido Y (NPY), noradrenalina (NA) y Adenosin trifosfato (ATP) (17). La vía parasimpáticas que proviene del ganglio esfenopalatino y otico donde se ven involucrados el péptido intestinal vasoactivo (VIP), el péptido histidina isoleucina (PHI), la acetilcolinesterasa (AcE), el péptido histidina metionina 27 (PHM) y el péptido activador de la ciclasa pituitaria (PACAP). Por último, las fibras sensitivas que provienen del ganglio trigémino donde se encuentra la sustancia P (SP), el péptido relacionado con el gen de la calcitonina (PRGC), la neurocinina A (NKA), y el PACAP. A pesar de lo anterior, hay que aclarar que los neuropéptidos relacionados con la fisiopatología de la migraña, aunque están involucrados con ésta, no todos son vías importantes para el desarrollo de la enfermedad (20).
Teniendo en cuenta el efecto iniciador de la migraña, es decir, la depresion cortical diseminada, que probablemente es causada por una alteración de la permeabilidad neuronal y tiene una disfunción posterior de canales iónicos, es importante decir que también existen dos mecanismos neuronales subyacentes: la sensibilización periférica y la sensibilización central (19). Las estructuras sensitivas intracraneana reciben inervación de ramas del trigémino estimuladas por liberación neuronal y endotelial, abriendo receptores en las terminales neurogénicas. Esto lleva al incremento de la concentración de calcio intracelular, seguido de fosforilación y activación de proteincinasa (PKC) y tirosincinasa (TryK) que, a su vez, por mecanismos moleculares aumenta la liberación de sustancias como la CGRP y VIP y promueve la inflamación neurogénica. Este proceso conduce a la sensibilización periférica dada por la disminución del umbral de respuesta de las fibras meníngeas, potenciándose el dolor y la inflamación (21).
Las concentraciones de serotonina, la cual se forma a partir del L-triptófano, son altas en las plaquetas y en el tracto gastrointestinal, en el que las células enterocromafines poseen el 90% de los niveles de 5 Hidroxitriptamina (5HT) del cuerpo, y este hecho adquiere crucial importancia ya que la serotonina viaja por el torrente sanguíneo hasta el cerebro donde sufre procesos de hidroxilacion y descarboxilacion para realizar sus funciones a nivel central (22). Las neuronas ricas en esta sustancia se encuentran alrededor del tallo cerebral y de la formación reticular en especial en el núcleo caudalis del trigémino donde tienen la mayor concentración de serotonina, lo cual será un factor especial para desencadenar el proceso de activación del sistema trigémino vascular en el ataque de migraña. Las neuronas ricas en esta sustancia se encuentran alrededor del tallo cerebral y de la formación reticular; aunque son pocas, están en mayor concentración en cada región cerebral donde se ubican. Los receptores especiales de 5HT, al activarse, incrementan la hidrólisis de inositol fosfato y generan un aumento de la concentración de calcio en el proceso migrañoso.
Se han evidenciado niveles de 5HT disminuidos un 30% en plaquetas y un 60% en el plasma cuando ocurre la crisis de migraña. En general, los autores consideran que durante la crisis migrañosa hay una liberación de serotonina desde las neuronas y las plaquetas mediadas por receptores 5HT2B (22). De igual forma, además de la serotonina existen diferentes sustancias involucradas en el desarrollo de la cefalea como los receptores dopaminérgicos y la histamina, que tienen efectos vasculares de vasoconstricción o vasodilatación. Estudios recientes sugieren que existe una hipersensibilidad dopaminérgica en la migraña y en lo que respecta a la histamina, se ha visto incrementada a nivel plasmático por una posible liberación a partir de los leucocitos o por la activación de células mastocitarias. Aunque la histamina no cruza la barrera hematoencefalica esta actúa a través de receptores H1 que están en el endotelio (23).
Con respecto al PRGC involucrado claramente en la cascada migrañosa, se ha evidenciado que se encuentra distribuido en sistema nervioso central y periférico; se encuentra en las fibras no mielinizadas tipo C, y mielinizadas Agama asociado con los vasos sanguíneos (24). Las neuronas que contienen PRGC inervan los vasos sanguíneos cerebrales y la liberación de esta se produce por respuesta a estímulos físicos, químicos y mecánicos.
Estudios recientes revelan que el PRGC puede ser un marcador para la migraña crónica ya que se han encontrado anticuerpos en los vasos intracraneales y el ganglio trigémino, estructuras que conservan altas concentraciones de este péptido; se evidenciaron incrementos a nivel plasmático durante eventos migrañosos y sustancias como el sumatriptán puede ser el abortador del incremento del péptido y, por supuesto, causante de la disminución de la cefalea (25).
HIPOTESIS GLUTAMATÉRGICA
El glutamato es el neurotransmisor encargado principalmente de la excitabilidad en el sistema nerviosos central y se ha visto aumentado en un ataque de migraña. Al inicio de la serie de eventos de la cascada fisiopatológica hay un estado de hiperexcitabilidad en la corteza neuronal que ocasiona el aumento del flujo sanguíneo regional para compensar las demandas metabólicas. Posteriormente, se inicia una onda de despolarización neuronal como reflejo de una incapacidad del tejido para mantener esa actividad aumentada (26), esta onda daría los síntomas positivos del aura, la depresión cortical dejaría tras de si una zona despolarizada lo cual necesita de un flujo sanguíneo menor para suplir las demandas metabólicas. La zona que previamente se despolarizó reduciría la actividad neuronal, lo que puede ocasionar los síntomas negativos que durarían menos de 60 minutos, tiempo suficiente para la re polarización neuronal (27).
Finalmente, las sustancias químicas implicadas en la propagación de la depresión cortical como la sobreexpresión del glutamato, producirían una inflamación neurogénica de los vasos meníngeos lo que ocasionaría la activación del sistema trigémino vascular produciendo la cefalea característica de la migraña (28, 29).
DEFICIENCIA DE MAGNESIO
Esta teoría propone que la deficiencia de magnesio en el cerebro desencadena una secuencia de eventos que puede comenzar con la agregación de las plaquetas y la liberación de glutamato y, terminar con la liberación de 5 hidroxitriptamina, un potente vasoconstrictor. En los estudios clínicos, el magnesio oral ha mostrado beneficios para el tratamiento preventivo de la migraña y el tratamiento intravenoso con magnesio puede ser eficaz para el tratamiento agudo de la migraña, en particular en ciertos subgrupos de pacientes. Sin embargo, aún no se ha logrado obtener una evidencia clara acerca de la eficacia del tratamiento del ataque agudo de migraña con magnesio oral o IV (30).
MIGRAÑA EPISÓDICA Y MIGRAÑA CRÓNICA
Por muchos años se ha discutido la relación entre la migraña episódica (ME) y la migraña crónica (MC); desde el punto de vista fisiopatológico ambas presentan diferencias en cuanto a la prevalencia, la duración de los síntomas y el número de crisis. No obstante, estudios epidemiológicos demuestran una similitud en cuanto a los síntomas presentados y a la fisiopatología que las desencadena. Así mismo los estudios muestran una diferencia importante en cuanto a la calidad de vida, ausentismo escolar y/o laboral, siendo mayor en la MC (Tabla 2) (31).
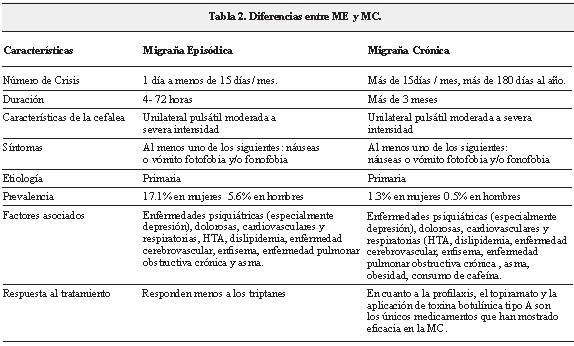
La ME está relacionada con un número de crisis igual a un día o menor de 15 días al mes, con una duración de 4 a 72 horas. En contraste, la MC presenta un número de crisis mayor a 15 episodios al mes o más de 180 días al año, con un periodo de duración de más de 3 meses. Buse menciona que la MC es una complicación de la ME (32), y muestra una prevalencia de aproximadamente 1.3 a 2.4% y demostró ser el desorden común más visto en la práctica clínica. Así mismo, Volcy publica en Acta Neurológica Colombiana en el 2013 una revisión de la fisiopatología de la migraña crónica en la que plantea los mecanismos de sensibilización central y periférica para que se produzca dicha cronificación del dolor y resume los factores de riesgo para este fenómeno (33). En su revisión establece comorbilidades que podrían explicar la predisposición del sujeto para desarrollar ME, entre los que se incluyen trastornos neurológicos (por ejemplo, ACV y epilepsia), trastornos psiquiátricos (ansiedad, depresión y trastorno de pánico), dolor crónico y otros trastornos como el asma y la enfermedad coronaria.
En contraste, las comorbilidades de MC rara vez se han estudiado en la población; sin embargo, se encontró en un artículo de revisión que hay factores que predisponen a la cronificación de la migraña como la obesidad con un IMC >30, el número de crisis migrañosas (a mayor número de crisis, mayor riesgo de desarrollar migraña crónica), la cafeína, trastornos del sueño, síndrome de Apnea/Hipopnea obstructiva del sueño ((SAHOS) en el que se propone que ocurre por fluctuaciones en la presión intracraneal y arterial durante el ronquido en un paciente susceptible a progresión del dolor e hipoxia), alodinia cutánea (marcador clínico de sensibilización en las neuronas de segundo orden), depresión, trastorno del pánico y ansiedad. Por lo anterior no se puede explicar claramente si hay o no una relación entre ambas patologías, pero sí podemos encontrar una clara relación entre estos factores predisponentes y el desarrollo de una ME o una MC como lo menciona Buse en su importante artículo (32).
Desde el punto de vista fisiopatológico, se han encontrado múltiples áreas cerebrales involucradas en el procesamiento del dolor durante la migraña. Este se ha relacionado con varios cambios funcionales y estructurales de diferentes áreas que pueden estar en relación con los procesos de cronificación de la enfermedad. Tales procesos involucran factores genéticos, ambientales, hormonales, neurobiológicos entre los que se encuentran: disfunción glutamatérgica del sistema trigémino vascular, del sistema nervioso autónomo, y de la liberación de neurotransmisores (34). En relación con los factores genéticos, Sobrino resume los estudios en los que se demuestra los genes implicados en la migraña hemipléjica familiar (35), aunque previamente, en el 2008, Isaza y colaboradores caracterizaron los conglomerados de clases latentes de la migraña familiar en un aislado genético en Antioquia (36).
A medida que pasa el tiempo y hay mayor exposición del cerebro a estímulos dolorosos (que podrían ser explicados por múltiples crisis de una ME y sumado a la persistencia de la enfermedad) se han visto cambios neuroplásticos mal adaptativos de la estructura y del funcionamiento cerebral que promueven la transformación a una MC. Estos cambios están relacionados con anormalidades metabólicas en estructuras del tallo cerebral, que se han postulado como generadores de la migraña. Entre estas se cuentan las siguientes estructuras: sustancia gris periacueductal (disminución de densidad neuronal), el núcleo cuneiforme, el hipotálamo, el tálamo y la corteza (cíngulo, ínsula, corteza prefrontal, corteza temporal y occipital), lo que lleva a un procesamiento anormal de los circuitos cerebrales (31). En todo este proceso patológico se desarrolla una lesión tisular con libración de mediadores inflamatorios (prostaglandinas E, FNT, bradicinina) con la posterior activación de canales iónicos y receptores que finalmente modifican los umbrales de respuesta y causan un aumento en la sensibilidad e hiperexcitabilidad neuronal (29).
CONCLUSIONES:
A pesar de que aún no se comprende con exactitud cuáles son los factores disparadores de la crisis de migraña ni las razones para que el dolor se vuelva crónico en algunos pacientes y en otros mejore con el paso del tiempo, las investigaciones actuales plantean que la teoría vascular de la migraña no aclara la cascada de eventos existentes en el proceso y que se debe recurrir a múltiples hechos que expliquen finalmente el dolor. Entre las explicaciones alternativas se encuentra la activación del sistema trigémino vascular como el principal factor desencadenante para desarrollar migraña con aura.
Se sigue considerando que no hay una sola hipótesis que explique el conjunto de factores implicados en la cascada de acontecimientos disparadores de la migraña. Por tanto, se deben estudiar otros mecanismos que adquieren mayor peso en la evidencia disponible, en los que la sensibilización central, la alodinia, el péptido relacionado con el gen de la calcitonina y el glutamato son los elementos con mayor proyección tanto para explicar el mecanismo fisiopatológico exacto, como para futuras posibilidades en el manejo farmacológico y no farmacológico de esta patología. Esto, dado que en muchas ocasiones es objeto de un enfoque terapéutico pobre en el que se minimiza el impacto de la carga de discapacidad que puede generar en pacientes jóvenes. Así mismo, obliga a replantear el abordaje y conocimiento que se tiene sobre las teorías fisiopatológicas de la migraña e incluso revisar los conceptos y competencias clínicas que se enseñan a las nuevas generaciones de estudiantes de pre y post grado.
Conflicto de intereses. Los autores declaran no tener conflicto de intereses.
Referencias
1. GOADSBY P, OSHINSKY M. Pathophysiology of Headache. In Wolff s Hedadache and Other Head Pain. Silberstein S, Lipton RB, Dodick DW eds. 8Th Edition. New York: Oxford University Press; 2008. 7; p. 105-119. [ Links ]
2. LANCE JW, GOADSBY PJ. Mechanism and management of headache. 7th edition. New York: Elsevier; 2005. [ Links ]
3. FERRARI MD, GOADSBY PJ. Migraine as a cerebral ionopathy with abnormal central sensory processing. In: Gilman S, editor. Neurobiology of disease. New York: Elsevier; 2007. p. 333-48. [ Links ]
4. SILBERSTEIN SD, LIPTON RB, GOADSBY PJ. Headache in clinical practice. 2nd edition. London: Martin Dunitz; 2002. [ Links ]
5. JOUTEL A, DUCROS A, VAHEDI K, ET AL. Genetic heterogeneity of familial hemiplegic migraine. Am J Hum Genet 1994; 55:1166-72. [ Links ]
6. OPHOFF RA, RV EIJK, SANDKUIJL LA, ET AL. Genetic heterogeneity of familial hemi- plegic migraine. Genomics 1994; 22:21-6. [ Links ]
7. KRUIT MC, LAUNER LJ, FERRARI MD, VAN BUCHEM MA. Brain stem and cerebellar hyperintense lesions in migraine. Stroke 2006; 37:1109-12. [ Links ]
8. NOZARI A, DILEKOZ E, SUKHOTINSKY I, ET AL. Microemboli may link spreading depression, migraine aura and patent foramen ovale. Ann Neurol 2009; published online Sept 14. DOI:10.1002/ana.21871. [ Links ]
9. TIETJEN GE, AL-QASMI MM, ATHANAS K, DAFER RM, KHUDER SA. Increased von Willebrand factor in migraine. Neurology 2001; 57:334-36. [ Links ]
10. CESAR JM, GARCIA-AVELLO A, VECINO AM, SASTRE JL,ALVAREZ-CERMENO JC. Increased levels of plasma von Willebrand factor in migraine crisis. Acta Neurol Scand 1995; 91:41-13. [ Links ]
11. BARTSCH T, LEVY MJ, KNIGHT YE, ET AL. Differential modulation of nociceptive dural input to [hypocretin] Orexin A and B receptor activation in the posterior hypothalamic area. Pain 2004;109:367-78. [ Links ]
12. SCHOONMAN GG, VAN DER GROND J, KORTMANN C, ET AL. Migraine headache is not associated with cerebral or meningeal vasodilatation-a 3T magnetic resonance angiography study. Brain 2008;131:2192-200. [ Links ]
13. AMIN FM, ASGHAR MS, HOUGAARD A, ET AL. Magnetic resonance angiography of intracranial and extracranial arteries in patients with spontaneous migraine without aura: a cross-sectional study. Lancet Neurol 2013; 12:454-61. [ Links ]
14. HOLLAND P, GOADSBY P. The hypothalamic orexinergic system: Pain and Primary Headaches. Headache-Current Headache 2007:951-962. [ Links ]
15. OLESEN J, BURSTEIN R, ASHINA M, TFELT-HANSEN P. Origin of pain in migraine: Evidence of peripheral sensitization. Lancet Neurol 2009; 9:679- 690. [ Links ]
16. WAEBER C. Serotonin and other biogenic Amines. En Olesen J, Goadsby PJ, Ramadan NM, Tfelt-Hansen P, Welch KWA eds. The Headaches, 3rd Ed. Philadelphia: Lippincott-Williams and Wilkins; 2006. Chapter 14; p. 143-149. [ Links ]
17. HAMEL E. Serotonin and migraine: Biology and Clinical implications. Cephalalgia- Headache Currents. 2007; 27:1295-1300. [ Links ]
18. ZARCO LA, PRETELT F, MILLAN S. Sistema Trigémino Vascular y Cefalea. Univ. Med 2013; 54(1): 92-103. [ Links ]
19. AFRIDI SK, GIFFIN NJ, KAUBE H, FRACKOWIAK RS, GOADSBY PJ. A PET study in spontaneous migraine. Arch Neurol 2005; 62: 1270-1275. [ Links ]
20. GUPTA S, NAHAS SJ, PETERLIN L. Chemical mediators of migraine: Preclinical and clinical observations. Headache 2011; Headache Currents: 1029- 1045. [ Links ]
21. JOHNSON KW, BOLAY H. Neurogenic inflammatory mechanisms. In Olesen J, Goadsby PJ, Ramadan NM, Tfelt-Hansen P, Welch KWA eds. The Headaches, 3rd Ed. Philadelphia: Lippincott-Williams and Wilkins; 2006. Chapter 33; p. 309-319. [ Links ]
22. GEPPETTI P, ROSSI E, CHIARUGI A, BENEMEI S. Antidromic vasodilation and the migraine mechanism. J Headache Pain 2012; 13:103-111. [ Links ]
23. PALM-MEINDERS IH, KOPPEN H, TERWINDT GM, ET AL. Structural brain changes in migraine. JAMA 2012; 308:1889-97. [ Links ]
24. BORSOOK D, MALEKI N, BECERRA L, ET AL. Understanding migraine through the lens of maladaptive stress responses: a model disease of allostatic load. Neuron 2012; 73:219-34. [ Links ]
25. EIKERMANN-HAERTER K, LEE JH, YUZAWA I, ET AL. Migraine mutations increase stroke vulnerability by facilitating ischemic depolarizations. Circulation 2012; 125:335-45. [ Links ]
26. MESSINA, MARIA A ROCCA, Cortical abnormalities in patients with migraine: a surface- based analysis. Radiology 2013; 268(1): 170-180. [ Links ]
27. BAHRA A, MATHARU MS, BUCHEL C, FRACKOWIAK RS, GOADSBY PJ. Brainstem activation specific to migraine headache. Lancet 2001; 357:1016-7. [ Links ]
28. MARK A LOUTER, JOHANNEKE E BOSKER, WILLEBRORDUS P J VAN OOSTERHOUT. Cutaneous allodynia as a predictor of migraine chronification. Brain 2013: 136: 3489 - 3496. [ Links ]
29. VOLCY M, Fisiopatología de la migraña, Acta Neurol Colomb 2013;29:44-52. [ Links ]
30. SUNEDELSTEIN C, MAUSKOP A. Role of magnesium in the pathogenesis and treatment of migraine. Expert Rev Neurother. 2009; 9(3):36979. [ Links ]
31. TIETJEN GE Migraine as a systemic vasculopathy. Cephalalgia 2009; 29:989-96. [ Links ]
32. BUSE DC, MANACK A, SERRANO D, TURKEL C, LIPTON RB. Sociodemographic and comorbidity profiles of chronic migraine and episodic migraine sufferers. J Neurol Neurosurg Psychiatry February 17, 2010. [ Links ]
33. VOLCY M. Pathophysiology of chronic migraine. Acta Neurol Colomb 2013;29:1 (Supl 1:1)25-30). [ Links ]
34. Martins Oliveira A, Speciali JG, Dach F, Marcaccini AM, Gonçalves FM, Gerlach RF, et al. Different circulating metalloproteinases profiles in women with migraine with and without aura. Clin Chim Acta. 2009, Oct;408(12): 604. [ Links ]
35. SOBRINO F, RAMOS M. Factores genéticos de la migraña. Acta Neurol Colomb 2013; 29:1 (Supl 1:1): 31-42. [ Links ]
36. ISAZA R, PINEDA D, AGUIRRE D, GALLEGO C. Caracterización clínica y de conglomerados de clases latentes de la migraña familiar en el aislado genético de Antioquia. Act Neurol Colomb 2008; 24:24-32. [ Links ]
