










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.3 Bogotá July/Set. 2015
Caso Clínico
Forma esporádica de enfermedad de Creutzfeldt-Jakob: reporte de dos casos
Sporadic Creutzfeldt-Jakob disease: two case reports
Guillermo González M. (1), Ángel Galván Marín (2), Nicolás Vargas P. (3), Cindy Beltrán E. (4), Efraín Amaya V. (5)
(1) Neurólogo Clínico, jefe Unidad de Neurología, Hospital Universitario Hernando Moncaleano Perdomo, Neiva (Huila). Docente de planta, Departamento de Ciencias Clínicas, Universidad Surcolombiana, Neiva, Huila, Colombia. Investigador adscrito al Grupo Dneuropsy.
(2) Médico General, Universidad Antonio Nariño, Bogotá, Colombia. Residente III año de Medicina Interna, Universidad Surcolombiana, Neiva, Huila, Colombia.
(3) Médico General, Universidad Surcolombiana, Neiva, Huila, Colombia. Investigador adscrito al Grupo Dneuropsy.
(4) Residente II año de Medicina Interna, Universidad Surcolombiana, Neiva, Huila, Colombia.
(5) Neurólogo Clínico, Hospital Universitario Hernando Moncaleano Perdomo, Neiva, Huila, Colombia. Docente de planta, Departamento de Ciencias Clínicas, Universidad Surcolombiana, Neiva. Investigador adscrito al Grupo Dneuropsy.
Recibido: 21/03/15. Aceptado: 10/08/15.
Correspondencia: Guillermo González: feevon@hotmail.es
Resumen
La enfermedad de Creutzfeldt-Jakob es la encefalopatía espongiforme más común en el ser humano y prototipo de las patologías causadas por priones. Se caracteriza histológicamente por astrogliosis y degeneración de la sustancia gris. Típicamente inicia con síntomas prodrómicos no específicos progresando a demencia con mioclonias y ataxia. Presentamos dos casos de mujeres en edad media con deterioro cognitivo progresivo, dificultades motrices, alteraciones del lenguaje y mioclonias que conducen a la muerte. En electroencefalogramas de ondas trifásicas lentas periódicas así como elevación de proteínas tau y 14-3-3 en LCR por apoyo del The National Prion Disease Pathology Surveillance Center - Cleveland, todos estos hallazgos definen las condiciones para el diagnóstico clínico de enfermedad por priones. El diagnóstico diferencial en el contexto de demencia rápidamente progresiva es amplio, incluyendo infecciones, intoxicaciones, trastornos metabólicos, autoinmunidad, vasculopatías y neoplasias que podrían explicar un posible subregistro en las estadísticas regionales. Existe una posible asociación de riesgo entre enfermedad por priones y médicos patólogos que, aunque discutida, podría limitar el estudio de los especímenes histológicos que son la clave del diagnóstico definitivo. A pesar de la importancia en salud pública de estas condiciones, el actual modelo de salud limita el manejo integral de los pacientes.
Palabras clave: Astrogliosis, Creutzfeldt-Jakob, demencia, mioclonia, priones, proteína 14-3-3 (DECS).
Summary
Creutzfeldt-Jakob is the most common spongiform encephalopathy in humans and the prototype of prions diseases. Astrogliosis and degeneration of the gray matter are the histological features. Typically starts with nonspecific prodromal symptoms that progressing to dementia with myoclonus and ataxia. We present two cases of women in middle age with progressive cognitive impairment, motor difficulties, language disorders and myoclonus that lead to death. EEG slow periodic triphasic waves and elevated protein tau and CSF14-3-3 support for The National Prion Disease Pathology Surveillance Center - Cleveland, all these findings define the conditions for the clinical diagnosis of prion disease. The differential diagnosis in the context of rapidly progressive dementia is broad including infections, poisoning, metabolic disorders, autoimmunity, vascular disease and neoplasms that could explain a possible underreporting in regional statistics. There is a possible risk association between disease and Medical Pathologists that although discussed could limit the study of histological specimens that are key to definitive diagnosis. Despite the public health importance of these conditions the current model of health limits the comprehensive management of patients.
Key words: Astrogliosis, Creutzfeldt-Jakob, dementia, myoclonus, prions, protein 14-3-3 (MeSH).
Introducción
A comienzos del siglo XX, de manera independiente, los neurólogos alemanes Hans-Gerhard Creutzfeldt y Alfons Maria Jakob describieron casos de demencia rápidamente progresiva y mioclonias que llevan inexorablemente a la muerte (1). En los análisis encefálicos post mórtem primaban los hallazgos de astrogliosis y degeneración de la sustancia gris, patrón que se encontraría repetitivamente en los casos subsecuentes.
En los años cincuenta Gajdusek y Blumberg describieron el kuru, una enfermedad neurodegenerativa endémica en las tribus de Nueva Guinea por la ingestión de tejidos cerebrales en ritos funerarios. Aunque Gajdusek lo atribuyó a un "virus lento", su descripción innovadora lo hizo merecedor, junto a su colaborador, del Premio Nobel de Medicina, en 1976 (2).
Años más tarde se definió que este prototipo de enfermedades son causadas por priones (proteinatious infectious particle that lacks nucleic acid), término que introdujo Stanley Prusiner (3) en la literatura médica para referirse a condiciones neurodegenerativas fatales causadas por partículas infecciosas no habituales (sin ácido nucleico). Este mecanismo, que involucra la infección por un agente distinto a los patógenos clásicos (bacterias, virus, hongos o parásitos), le acreditó a Prusiner en 1997 el Premio Nobel de Medicina (4) y lo constituyó en un desafío para el dogma actual de la biología molecular y como materia de intensa investigación en la actualidad.
El mecanismo fisiopatológico de la enfermedad de Creutzfeldt-Jakob no ha sido dilucidado en su totalidad, pero hay descripciones que lo pueden explicar en parte. Kristiansen et al. (5) demostraron experimentalmente con murinos que las enfermedades neurodegenerativas caracterizadas por depósitos intracelulares de diferentes sustratos se deben a la reorganización neuronal inducida por una proteína anormal secundaria a la alteración de la actividad de la vía proteasoma-ubiquitina S26, elemento básico para la degradación de proteínas y sustratos neurotóxicos, lo que conlleva finalmente a la pérdida de neuronas, formación anómala de vacuolas intracelulares, acúmulos de glías/astrocitos y formación de espacio en el parénquima cerebral como la característica de encefalopatía espongiforme.
La enfermedad de Creutzfeldt-Jakob es la encefalopatía espongiforme más común en el ser humano. Típicamente inicia con síntomas prodrómicos no específicos (astenia, cefalea, alucinaciones, cambios en el comportamiento, sueño y apetito) progresando finalmente a demencia y típicamente mioclonias/ataxia en el 80% de los casos (6). Aunque más raras, están descritas otras cuatro variantes clínicas: variante de Oppenheimer relacionada principalmente con compromiso atáxico severo, variante de Heidenhain de compromiso cortical (visión borrosa, alucinaciones visuales y ceguera), variante pancencefalítica y variante amiotrófica (7). Recientemente se ha identificado la enfermedad de Creutzfeldt-Jakob como una de las causas etiológicas de los estados epilépticos no convulsivos, en especial aquellos de difíciles manejos refractarios al uso de múltiples anticonvulsivantes y sedantes en los cuales ya se han excluido otras causas más comunes (8). También se han descrito pacientes en quienes esta patología puede debutar con síntomas depresivos severos de rápida evolución, llevando incluso al síndrome catatónico (9).
Las lesiones histopatológicas de localización típica en ganglios basales, tálamo, cerebelo y corteza cerebral se resumen en pérdida neuronal, gliosis y degeneración espongiforme (vacuolas de múltiples tamaños) (10), confiriendo el denominativo "encefalopatía espongiforme transmisible".
Caso 1
Paciente femenina de 48 años, natural de Líbano (Tolima), residente en Neiva (Huila), ocupación: asesora comercial, divorciada, con tres hijos sanos. Valorada por consulta externa de neurología por cuatro meses de evolución de pérdida de la memoria, dificultades motrices, hipoestesias en cabeza, manos y pies, que en ocasiones no responde al llamado, con episodios de lenguaje incoherente. Se decide hospitalizar para ampliación de estudios diagnósticos. Como antecedentes positivos se registraron cistopexia, histerectomía y tonsilectomía. Todos los estudios paraclínicos iniciales fueron negativos (Tabla 1). Resonancia magnética cerebral inicial dentro de límites normales. Electroencefalograma inicial evidencia actividad epiléptica manejada con dosis única de midazolam intravenoso y posterior deterioro del patrón respiratorio, requiriendo traslado a unidad de cuidado intensivo. Como diagnóstico probable se plantea encefalitis límbica, solicitándose PET con turno en la Fundación Santa Fe (Bogotá), pero no fue posible debido a estatus epiléptico e imposibilidad de traslado. Con la finalidad de descartar enfermedad maligna oculta se realizan ecografía mamaria, con hallazgo de enfermedad fibroquística; ecografía transvaginal normal y TAC de tórax simple/contrastado normal. Presenta episodios febriles, documentándose neumonía aspirativa, tratada con ampicilina sulbactam. Se realiza salpingooforectomía bilateral más liberación de adherencias peritoneales, tras lo cual no hay recuperación neurológica.
Egresa de UCI con importante compromiso neurológico, alerta, pero no obedece ni responde órdenes, con espasticidad de extremidades y mioclonias con estímulos auditivos.
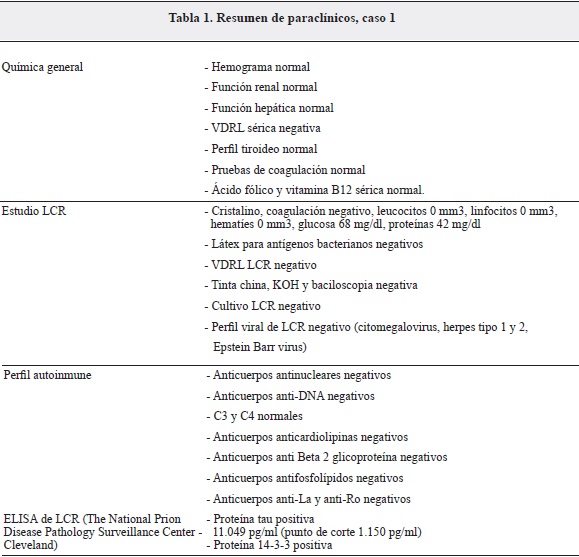
Durante hospitalización complicada fue tratada por bacteriemia de origen en vías urinarias. En electroencefalograma de control se encuentra patrón de ondas trifásicas de punta - onda aguda lenta en forma periódica menor de dos segundos, característico de enfermedad de Creutzfeldt-Jakob (Figura 1).
Por clínica y hallazgos en EEG se solicita proteína 14-3-3 en líquido cefalorraquídeo (se hace requerimiento a centros académicos norteamericanos de enfermedades por priones).
En cuanto a los diagnósticos diferenciales se solicitaron anticuerpos para rabia en LCR (enviados al Instituto Nacional de Salud) que fueron negativos. Se solicitó valoración por toxicología para descartar intoxicación por metales pesados y se conceptúa baja probabilidad, se solicitaron muestras que nunca fueron tomadas debido a no aprobación por parte de la entidad promotora de salud de la paciente.
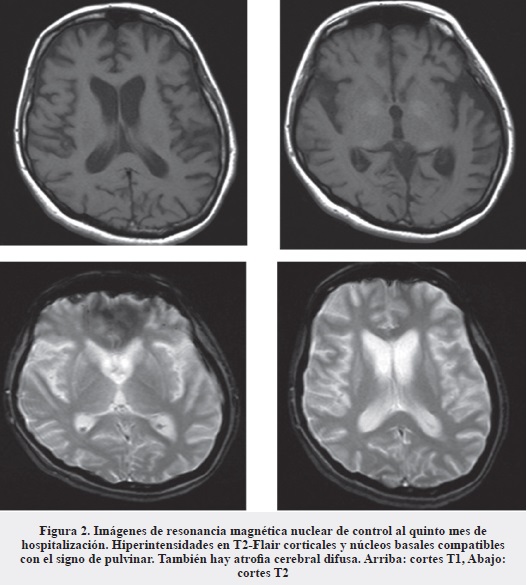
La evolución continúa estacionaria; se solicita nueva RMN cerebral simple y contrastada que muestra hiperintensidades en T2-Flair corticales y núcleo basales, secuelas de probable injuria vascular en núcleo basal derecho, así como atrofia cerebral difusa (Figura 2).
Se recibe reporte del The National Prion Disease Pathology Surveillance Center -Cleveland, informando que se realizó análisis por Elisa en la muestra enviada de LCR, encontrando un valor positivo de 11.049 pg/ml para la proteína tau (punto de corte 1.150 pg/ml) y proteína 14-3-3 positiva. Ofrecen estudio post mórtem del tejido cerebral de manera gratuita.
Se reporta el caso a la Secretaría de Salud del Huila mediante ficha epidemiológica y se refuerzan las medidas de aislamiento de la paciente. Se inicia la gestión para que una vez ocurra el deceso se proceda a la extracción del encéfalo bajo las condiciones solicitadas por el Instituto Nacional de Priones, así como el procesamiento y envío de las muestras para estudios patológicos. Se establece que en conjunto la Secretaría de Salud y el Hospital Universitario se encargarían del procedimiento por tratarse de un evento de interés en salud pública.
Alrededor de un mes después de estos eventos la paciente presenta deterioro rápido de su estado general, séptica, con aumento del trabajo respiratorio y desaturacion progresiva que la llevan a la muerte. La Secretaría de Salud autoriza la inhumación del cadáver sin la realización de los estudios de necropsia.
Caso 2
Paciente femenina de 55 años, natural y residente en Neiva (Huila), ama de casa y casada. Ingresa al servicio de psiquiatría y medicina interna por cuadro clínico de dos meses de evolución de trastornos de la memoria reciente, alteración del comportamiento e insomnio, que se relacionan con la muerte de la madre.
Cinco días previos al ingreso se torna agresiva, coprolálica, ideas celotípicas con el esposo, hiporexia y emesis. Como antecedentes de importancia: hipotiroidismo y diabetes mellitus tipo 2, en manejo con insulina y metformina. Al examen físico, paciente alerta, deshidratada, con evidencia de lesión eritematosa en pie derecho que sugiere infección de tejidos blandos. Se consideró en un principio paciente con diabetes mellitus descompensada por pie diabético y reacción situacional, medicándose con risperidona, biperideno, haloperidol, insulinoterapia y antibiótico parenteral. En los estudios iniciales, hemograma, electrolitos, perfil tiroideo, función renal y función hepática normales, únicamente uroanálisis con glucosuria y glucometrías en rangos 200-300 mg/dl (Tabla 2).
En el quinto día de hospitalización la paciente presentó deterioro clínico dado por episodios de agitación psicomotora, rigidez, espasticidad generalizada, mioclonias y desconexión con el medio externo. En este momento es conocida por el servicio de neurología, realizándose TAC de cráneo simple normal, estudio de LCR normal y electroencefalograma que muestra estatus epiléptico no convulsivo manejado en la unidad de cuidados intensivos con infusión de midazolam y levetiracetam. El electroencefalograma de control a las veinticuatro horas muestra el típico patrón onda punta - onda aguda - onda lenta trifásica periódica (Figura 3). Teniendo en cuenta el contexto clínico de la paciente de demencia rápidamente progresiva con episodios de mioclonias y patrón electroencefalográfico típico, se enfoca como posible enfermedad de Creutzfeldt-Jakob. Se realiza RNM cerebral normal.
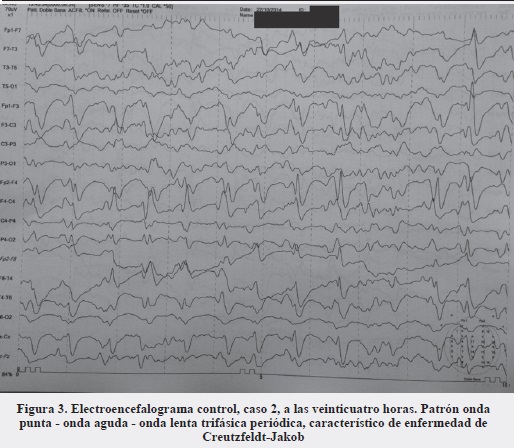
La paciente egresa de la unidad de cuidados intensivos luego de diez días estable hemodinámicamente, pero sin mejoría del estado neurológico. En el servicio de hospitalización su evolución es tórpida, complicándose con neumonía nosocomial grave que la lleva a la muerte.
Antes de la defunción se solicitó el análisis de líquido cefalorraquídeo, que se procesó en The National Prion Disease Pathology Surveillance Center - Cleveland, informando para la proteína tau por técnica de Elisa un valor positivo de 4.434 pg/ml (punto de corte 1.150 pg/ml), así como proteína 14-3-3 positiva. Este centro ofrece además el estudio post mórtem del tejido cerebral, que no se pudo realizar por dificultades técnicas en el envío de especímenes biológicos a otros países.
Discusión
Aunque la media de edad reportada en la literatura es de 68 años (11), nuestras pacientes son más jóvenes de lo que dicta esta tendencia estadística, destacando la importancia de sospechar el diagnóstico en el adulto joven.
La denominada "forma esporádica" es la presentación más común de enfermedad de Creutzfeldt-Jakob, que implica descartar el tipo familiar (autosómica dominante por mutaciones de proteínas priónicas cerebrales), forma variante (ingesta cárnica de ganado con encefalopatía espongiforme bovina) y la forma iatrogénica (contaminación con hemoderivados, trasplantes de córnea, injertos de duramadre, electrodos intracerebrales y extractos humanos de hormona de crecimiento).
Hay controversia respecto a la relación causal en la forma iatrogénica, dado que estas asociaciones están basadas en estudios de casos y controles con posibles sesgos en selección y comparación de los controles; así mismo, existe incertidumbre en la asociación con procedimientos dentales (12).
Los hallazgos electroencefalográficos consisten en ondas agudas trifásicas periódicas (13). En las secuencias T2 de la resonancia magnética los hallazgos típicos incluyen hiperintensidades simétricas bilaterales en los ganglios basales (signo del pulvinar), putamen y cabeza del caudado. También son frecuentes las lesiones de la corteza cerebral (14). El análisis de proteína 14-3-3 en líquido cefalorraquídeo tiene una sensibilidad reportada de 92% con especificidad variable según distintas series, pero especialmente baja (82-87%) en otros desórdenes como procesos neurodegenerativos, neoplasias, accidentes vasculares cerebrales, epilepsia y condiciones psiquiátricas (15). Según recomendación del Colegio Americano de Neurología, debe realizarse esta prueba diagnóstica en pacientes con demencia rápidamente progresiva, sospecha clínica de enfermedad de Creutzfeldt-Jakob e incertidumbre diagnóstica (16). El estándar de oro para el diagnóstico es la detección de proteínas priónicas (PrPSc) en biopsias cerebrales o autopsia.
En los últimos años las publicaciones han demostrado alta correlación de los hallazgos típicos de resonancia magnética, electroencefalograma y biomarcadores en líquido cefalorraquídeo en el diagnóstico pre mórtem de la enfermedad de Creutzfeldt-Jakob esporádica; la asociación de estos en el contexto de neurodegeneración rápida confiere el diagnóstico clínico (17).
Recientemente se están investigando métodos diagnósticos alternativos, detectando priones en muestras distintas de líquido cefalorraquídeo, como podría ser la mucosa olfatoria (18), pero surge la preocupación de una nueva forma iatrogénica de transmisión mediante los fibroscopios usados para la obtención de las muestras. Lo anterior podría superarse con la búsqueda de las proteínas priónicas en orina, como lo demuestran Moda et al. (19) en un reciente trabajo multicéntrico, con sensibilidad del 100% y especificidad de 92%. Cabe resaltar que el anterior trabajo fue realizado solamente con formas variantes y su significado en las otras presentaciones de la enfermedad es desconocido. De momento estas modalidades alternativas no se encuentran estandarizadas y no se recomienda su uso en general.
Creemos que las pacientes sufrieron formas esporádicas, dado que son casos índice en la región con familiares sanos, sin antecedentes personales que impliquen exposición a tejidos humanos, y sin casos de enfermedad de las "vacas locas" reportados por las autoridades sanitarias locales.
Se ha reportado la posible asociación entre formas variantes de enfermedad de Creutzfeldt-Jakob y profesionales de salud (específicamente patólogos) que por su labor están expuestos a tejidos humanos contaminados; esta situación, aunque debatida por grupos de expertos mundiales en neuroepidemiología (20), puede obstaculizar el estudio de un posible caso con las esperables consecuencias en salud pública local. Recomendamos las convencionales medidas de protección personal aceptadas universalmente para la manipulación de muestras diagnósticas, tejidos y fluidos de pacientes con sospecha de la enfermedad.
El diagnóstico diferencial es amplio y constituye un reto clínico; se incluyen demencias, infecciones (encefalitis posviral y encefalitis por HIV), intoxicaciones (litio/mercurio), trastornos metabólicos (principalmente alcohol), autoinmunidad (lupus, vasculitis primarias del SNC), trastornos vasculares ateroscleróticos y neoplasias (primarias y metástasis) (21-23). En el grupo de demencias rápidamente progresivas se incluyen la enfermedad de Alzheimer, demencia frontotemporal, enfermedades asociadas a mioclonias, demencia por cuerpos de Lewys y la parálisis supranuclear (24).
Aunque todas estas entidades tienen un deterioro neurocognitivo rápido, la enfermedad de Creutzfeldt-Jakob progresa en el curso de pocos meses, por lo cual enfatizamos el estudio cuidadoso de los pacientes con demencia rápidamente progresiva, especialmente pacientes jóvenes en quienes creemos podría existir un subdiagnóstico local.
A la fecha no existe tratamiento para ninguna forma de Creutzfeldt-Jakob, solo se puede ofrecer tratamiento paliativo, ya que la enfermedad sigue un curso progresivo con mortalidad del 100%. Por lo anterior, las enfermedades por priones adquieren especial importancia en salud pública; específicamente en Colombia, están reglamentadas las medidas de prevención y vigilancia de la forma variante, que son de obligatorio cumplimiento (25-27). Pese a que las enfermedades por priones son materia de intensa investigación, el conocimiento sigue siendo escaso. Esta entidad se constituye en un campo de vertiginoso desarrollo para las neurociencias en los próximos decenios, para lo cual el médico de todo nivel de atención debe estar preparado.
Finalmente, clasificamos nuestros casos como probables formas esporádicas de enfermedad de Creutzfeldt-Jakob (el caso definitivo lo determinaría el estudio patológico). Debido principalmente al actual modelo regente se fraguan innumerables e infranqueables barreras que limitan garantizar un servicio integral de salud a los pacientes.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Referencias
1. TRIARHOU LC, JAKOB AM (1884-1931). Neuropathologist par Excellence. Eur Neurol. 2009;61:52-8. [ Links ]
2. http://www.nobelprize.org/nobel_prizes/medicine/laureates/1976/ Consultado: julio 20 de 2015. [ Links ]
3. PRUSINER SB. Prions. Proc Natl Acad Sci USA 1998;95(23):13363-83. [ Links ]
4. http://www.nobelprize.org/nobel_prizes/medicine/laureates/1997/ Consultado: julio 20 de 2015. [ Links ]
5. KRISTIANSEN M, DERIZIOTIS P, DIMCHEFF DE, ET AL. Disease-associated prion protein oligomers inhibit the 26S proteasome. Mol Cell. 2007;26:175-88. [ Links ]
6. COLLINS SJ, LAWSON VA, MASTERS CL. Transmissible spongiform encephalopathies. Lancet 2004;363:51-61. [ Links ]
7. HEIDENHAIN A. Klinische und anatomische Untersuchungen Über eine eigenartige organische Erkrankung des Zentralnervensystems in Praesenium. Z Ges Neurol Psychiatry 1928;118:49-114. [ Links ]
8. ESPINOSA P, BENSALEM-OWEN M, FEE D. Sporadic Creutzfeldt-Jakob disease presenting as nonconvulsive status epilepticus case report and review of the literature. Clinical Neurology and Neurosurgery 2010;112:537-40. [ Links ]
9. MILANLIOGLU A, ET AL. Catatonic depression as the presenting manifestation of creutzfeldt-Jakob disease. J Neurosci Rural Pract. 2015 Jan-Mar;6(1):122. [ Links ]
10. DEARMOND SJ, PRUSINER SB. Prion protein transgenes and the neuropathology in prion diseases. Brain Pathol. 1995;5(1):77-89. [ Links ]
11. AGUZZI A, BAUMANN F, BREMER J. The prion's elusive reason for being. Annu Rev Neurosci 2008;31:439-77. [ Links ]
12. CUESTA J, RUIZ M, WARD H, CALERO M, SMITH A, ALONSO C, ET AL. Sensitivity to Biases of Case-Control Studies on Medical Procedures, Particularly Surgery and Blood Transfusion, and Risk of Creutzfeldt-Jakob Disease. Neuroepidemiology 2012;39:1-18. [ Links ]
13. VENNETI S. Prion Diseases. Clin Lab Med. 2010 Mar;30(1):293-309. [ Links ]
14. LIM T. Magnetic Resonance Imaging Findings in Bilateral Basal Ganglia Lesions. Ann Acad Med Singapore 2009;38:795-802. [ Links ]
15. STOECK K, SÁNCHEZ-JUAN P, GAWINECKA J, GREEN A, LADOGANA A, POCCHIARI M, ET AL. Cerebrospinal fluid biomarker supported diagnosis of Creutzfeldt-Jakob disease and rapid dementias: a longitudinal multicentre study over 10 years. Brain 2012:135;3051-061. [ Links ]
16. MUAYQIL T, GRONSETH G, CAMICIOLI R. Evidence-based guideline: Diagnostic accuracy of CSF 14-3-3 protein in sporadic Creutzfeldt-Jakob disease: Report of the Guideline Development Subcommittee of the American Academy of Neurology. Neurology 2012;79:1499-506. [ Links ]
17. ZERR I, KALLENBERG K, SUMMERS DM, ROMERO C, TARATUTO A, HEINEMANN U. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. Brain 2009:132;2659-68. [ Links ]
18. ORRÚ C, BONGIANNI M, TONOLI G, FERRARI S, HUGHSON A, GROVEMAN B, ET AL. A Test for Creutzfeldt-Jakob Disease Using Nasal Brushings. N Engl J Med. 2014;371:519-29. [ Links ]
19. MODA F, GAMBETTI P, NOTARI S, CONCHA-MARAMBIO L, CATANIA M, PARK K, ET AL. Prions in the Urine of Patients with Variant Creutzfeldt-Jakob Disease. N Engl J Med. 2014;371:530-9. [ Links ]
20. ALCALDE-CABERO E, ALMAZÁN-ISLA J, BRANDEL JP, BREITHAUPT M, CATARINO J, COLLINS S, ET AL. Health professions and risk of sporadic Creutzfeldt-Jakob disease, 1965 to 2010. Euro Surveill 2012;17(15):pii=20144. [ Links ]
21. PATERSON RW, TAKADA LT, GESCHWIND MD. Diagnosis and treatment of rapidly progressive dementias. Neurol Clin Pract. 2012 Sep;2(3):187-200. [ Links ]
22. GESCHWIND MD, SHU H, HAMAN A, SEJVAR JJ, MILLER BL. Rapidly progressive dementia. Ann Neurol. 2008;64(1):97-108. [ Links ]
23. PATERSON RW, TAKADA LT, GESCHWIND MD. Diagnosis and treatment of rapidly progressive dementias. Neurol Clin Pract. 2012 Sep;2(3):187-200. [ Links ]
24. GESCHWIND MD. Diagnosis and treatment of rapidly progressive dementias. Neurol Clin Pract. 2012 Sep;2(3):187-200. [ Links ]
25. https://www.invima.gov.co/images/stories/aliementos/decreto_2350_2004.pdf Consultado: febrero 14 de 2015. [ Links ]
26. http://www.ica.gov.co/getattachment/7108ded1-e068-4186-9e67-Ocd29ce708c2/3752.aspx Consultado: febrero 14 de 2015. [ Links ]
27. http://www.ins.gov.co/lineas-de-accion/Subdireccion-Vigilancia/sivigila/Protocolos%20SIVIGILA/PRO%20Creutzfeld.pdf Consultado: febrero 14 de 2015. [ Links ]
