










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.3 Bogotá July/Set. 2015
Caso Clínico
Déficit de pantotenato quinasa asociado a neurodegeneración. Reporte de un caso clásico y revisión de la literatura
Pantothenate kinase-associated neurodegeneration: A classical case report and literature review
Heidy Gómez Naranjo (1), Eugenia Espinosa García (2), Ángela Paredes (3)
(1) Pediatra, Neuróloga Pediatra, Universidad Militar Nueva Granada, Bogotá, Colombia.
(2) Pediatra, Neuróloga Pediatra, jefe del Programa de Posgrado en Neurología Pediátrica, Universidad Militar Nueva Granada, Bogotá, Colombia, Hospital Militar Central. Neuróloga Pediatra, Instituto de Ortopedia Infantil Roosevelt.
(3) Residente de III año de Neurología Pediátrica, Universidad Militar Nueva Granada, Bogotá, Colombia.
Recibido: 5/03/15. Aceptado: 31/08/15.
Correspondencia: Heidy Gómez Naranjo: heidyjohannag@gmail.com
Resumen
El déficit de pantotenato quinasa asociado a neurodegeneración (PKAN por su sigla en inglés) es una enfermedad neurodegenerativa poco frecuente que se caracteriza por disfunción extrapiramidal progresiva y acumulación de hierro en los ganglios basales. El signo clásico en la neuroimagen de "ojos de tigre" se ve en las imágenes ponderadas en T2 en la resonancia magnética cerebral. A continuación presentamos un caso clásico de la enfermedad en un niño que inicia con síntomas motores a los 5 años de edad y que fue estudiado en el Instituto de Ortopedia Infantil Roosevelt, con neuroimágenes típicas y confirmación de la mutación por estudio molecular.
Palabras clave: Déficit de pantotenato quinasa asociado a neurodegeneración, distonía, espasticidad, neuroimagen con "ojos de tigre" (DECS).
Summary
Pantothenate kinase-associated neurodegeneration (PKAN) is a rare neurodegenerative disease characterized by progressive extrapyramidal dysfunction and iron accumulation in the basal ganglia. The classic sign in neuroimaging of "eye of the tiger" is seen on T2-weighted magnetic resonance imaging scan. We present a classic case of the disease in a child who starts with motor symptoms at 5 years old and was studied at the Instituto de Ortopedia Infantil Roosevelt, with typical neuroimaging and confirmation of the mutation by molecular study.
Key words: Pantothenate kinase-associated neurodegeneration, neurodegeneration, dystonia, spasticity, neuroimaging with "eye of the tiger" (MeSH).
Introducción
El PKAN, conocido también como síndrome de Hallervorden-Spatz, fue descrito en 1922. Llamado también neurodegeneración por acumulación cerebral de hierro-1, es una enfermedad neurodegenerativa autosómica recesiva de baja prevalencia que suele debutar en las primeras décadas de la vida y se caracteriza por depósito de hierro en los ganglios basales, causando disfunción extrapiramidal progresiva y manifestaciones clínicas caracterizadas por distonía progresiva, alteración del habla, retinopatía pigmentaria y déficit cognitivo, aunque un 25% de los pacientes presentan curso atípico caracterizado por inicio más tardío (> 10 años), alteración del habla, trastornos psiquiátricos y una progresión gradual de la enfermedad (1) .
Presentación del caso clínico
Se trata de un escolar de sexo masculino, de 12 años de edad, fruto del primer embarazo, complicado por preeclampsia y nacido a término, presentó hipoglicemia transitoria e ictericia a los 2 días de vida, sin otras complicaciones. El padre cursa con ataxia espinocerebelosa y hay antecedentes de enfermedad psiquiátrica por línea materna. El niño es remitido por presentar a los 5 años un trastorno del movimiento con ataxia y distonía, asociado a espasticidad de predominio en miembros inferiores, y retardo del desarrollo psicomotor.
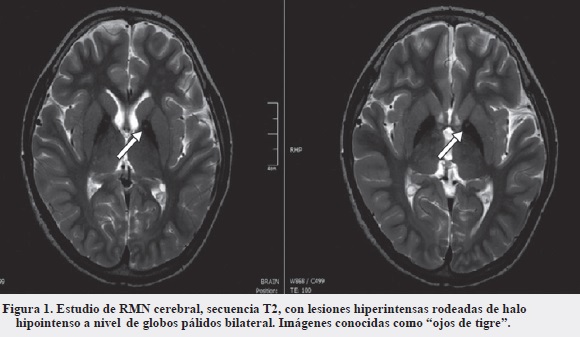
Al examen se evidenciaron movimientos distónicos generalizados e hiperreflexia musculotendinosa, trastorno del habla y déficit cognitivo. Se iniciaron estudios. Los ácidos orgánicos urinarios cualitativos, el perfil de acilcarnitinas y la relación ácido láctico/piruvato fueron normales; el estudio para Nieman Pick tipo C también fue negativo, la resonancia magnética cerebral inicial normal y el estudio molecular para atrofia espinocerebelosa fue negativo. El paciente presenta progresión de las distonías, con presencia de tormenta distónica en dos ocasiones. Se realizó manejo con múltiples fármacos, incluida bomba de baclofeno, sin mejoría. Se realiza resonancia magnética cerebral de tres teslas con cortes finos de ganglios basales, informándose hiperintensidad de globos pálidos bilateral, con halo de hipointensidad, imagen conocida como "ojos de tigre" que sugiere depósito de hierro (Figura 1).
Se solicitó el estudio molecular para déficit de pantotenato quinasa, reportando mutación en el gen PANK2 con cambio del nucleótido c1231 G-A a AA p.Gly411Arg homocigoto.
Con el estudio molecular, la clínica y resonancia magnética cerebral, se establece el diagnóstico de PKAN.
Neurodegeneración con acumulación de hierro cerebral
La neurodegeneración por acumulación de hierro cerebral (NBIA) corresponde a un grupo de trastornos genéticamente heterogéneos y progresivos. Se caracterizan por acumulación de hierro a nivel de ganglios basales y otras regiones del cerebro, y se manifiestan con movimientos extrapiramidales como parkinsonismo y distonía. La edad de inicio, gravedad y compromiso cognitivo son variables (2).
Las causas de NBIA pueden ser ocasionadas por varias causas. Alteraciones en la vía metabólica del hierro, desórdenes en el metabolismo de ceramidas/fosfolípidos, algunos desórdenes lisosomales en la regulación mitocondrial del hierro o desórdenes en la función de genes desconocidos (3, 4).
Entre los trastornos neurodegenerativos con acumulación de hierro cerebral está el PKAN, llamado neurodegeneración con acumulación cerebral de hierro-1 (NBIA1), que corresponde a los desórdenes en el metabolismo de ceramidas/fosfolípidos (5, 6).
Este trastorno presenta una imagen típica de "ojos de tigre" en la secuencia ponderada del T2 de la resonancia magnética cerebral. Sin embargo, este signo no es patognomónico de mutaciones en el gen PANK2 (5).
El síndrome de HARP (hipoprebeta- lipoproteinemia, acantocitosis, retinitis pigmentosa y degeneración palidal) es un trastorno poco frecuente y es considerado parte del espectro de la enfermedad PKAN pero con un fenotipo menos grave; este es causado por mutación del gen PANKA2 (1, 5).
Epidemiología
Se estima una prevalencia de 1 a 3 casos en cada 1.000.000, suponiendo un pequeño número de diagnósticos erróneos y casos perdidos (1, 6).
Se ha descrito una mutación fundadora común en los Países Bajos y en una comunidad al suroeste de República Dominicana: c.680A>G (p.Tyr227Cys) (1). La mayoría de los casos se presentan en la infancia o en el adulto joven (7).
Genética
El PKAN es causado por la mutación del gen PANKA2 situado en el locus 20p13, con variabilidad alélica (5).
El gen PANKA2 codifica para la quinasa mitocondrial pantotenato quinasa, enzima reguladora esencial en la biosíntesis de coenzima A (CoA) (5).
Fisiopatología
La enzima mitocondrial pantotenato quinasa regula la biosíntesis de la coenzima A o acil-CoA (CoA), catalizada desde la fosforilación citosólica de pantotenato (vitamina B5), N-pantotenoilcisteína y panteteína. La CoA interviene en la biosíntesis y oxidación de ácidos grasos, y descarboxilación oxidativa del ácido pirúvico, paso previo al ciclo de Krebs; el déficit causa daño en la vía metabólica de la síntesis de ácidos grasos y metabolismo energético. Además del déficit en la producción de CoA se acumula un producto tóxico: la cisteína (Figura 2). Esta quela el hierro, acumulándose en partes específicas del cerebro. Los sitios de mayor lesión es el cerebro, a nivel de ganglios basales, hipocampo, ciertos núcleos cerebelares (dentado), sustancia nigra, algunas regiones subcorticales y retina (4, 6-8).

El hierro es crítico para la embriogénesis y fisiología neural y participa en funciones celulares como citocinesis, mielinización, transporte de electrones (citocromo a, b, y c oxidasa), actividad de enzimas antioxidantes, metabolismo de aminas biogénicas, síntesis de lípidos, colesterol y sistema GABA (7).
El sistema nervioso central (SNC) tiene control en su microambiente y la barrera hematoencefálica, y se opone a la libre circulación de transferrina, ferritina y ceruloplasmina. El hierro se acumula en el SNC como función de la edad avanzada, y una porción del metal en el parénquima y líquido cefalorraquídeo (LCR) sigue siendo activo en procesos de oxidorreducción (redox), ocasionando daño en el funcionamiento cerebral (7).
En las NBIA, específicamente en PKAN, se altera la homeostasis del hierro, produciendo depósitos patológicos en el SNC con daños subsecuentes a las estructuras celulares mediante la participación en generación de especies oxígeno reactivas potencialmente neurotóxicas: producen reducción de H2O2 a OH (hidroxilo), llamada catálisis de Fenton, en la que el OH formado actúa como agente oxidante no selectivo muy potente que reacciona con compuestos orgánicos hasta la mineralización (9), o comportándose como actividad pseudoperoxidasa que bioactiva compuestos benignos en radicales intermedios tóxicos libres (7).
Las especies reactivas de oxígeno (ROS) producen: protoxinas bioactivas, señales celulares aberrantes, falla bioenergética, disfunción proteosomal, agregación de proteínas, formación de inclusiones, sinaptólisis, apoptosis y necrosis. El SNC de los mamíferos los hace vulnerables al hierro y sus daños redox son el flujo robusto de moléculas de oxígeno en la respiración celular, excesiva generación de especies oxígeno reactivas, susceptibilidad de SNC a la peroxidación lipídica, abundantes neurotransmisores oxidables (dopamina) y potencialmente excitotóxicos (glutamato) y escasez de ciertos antioxidantes (9).
Historia natural
PKAN clásico. Las características clínicas del PKAN clásico son homogéneas y se presentan en la primera infancia, por lo general antes de los 6 años de edad (media de 3,4 años) (1, 7). Los signos y síntomas neurológicos de inicio temprano son piramidalismo y síntomas extrapiramidales con distonía, disartria y rigidez (1, 7).
Es frecuente el compromiso distónico de las extremidades en forma asimétrica en los miembros inferiores, acompañados de corea o parkinsonismo (7), causando fracturas espontáneas de huesos largos por combinación de tensión ósea y osteopenia (1). El síntoma más común es el deterioro de la marcha, resultado de la combinación de rigidez de las extremidades, distonía y espasticidad. La distonía puede ocasionar trauma recurrente en la lengua y requerir extracción dentaria (1, 7).
Algunos niños presentan retraso en el desarrollo, siendo principalmente motor. La marcha en puntillas y posturas distónicas de las extremidades superiores son signos menos frecuentes (1). La distonía orofacial puede resultar en efectos secundarios por la dificultad para deglutir y desnutrición secundaria. La muerte prematura está relacionada con estos efectos secundarios (inmunodeficiencia relacionada con desnutrición, neumonía por aspiración) (1).
El compromiso del tracto corticoespinal es común e incluye espasticidad, hiperreflexia, y signo de Babinski (1, 7). Las convulsiones son raras, pero el deterioro intelectual es característico de PKAN, aunque algunos estudios muestran que el deterioro cognitivo puede ser sobreestimado debido a deficiencias motoras (1). Los pacientes pueden presentar también alteraciones psiquiátricas como trastorno obsesivo compulsivo, manías, psicosis y depresión (7).
Los síntomas visuales pueden ser los que llevan a los niños a consulta y presentar restricción de campos visuales secundarios a retinopatía; la degeneración pigmentaria retiniana se presenta en 2/3 partes de los pacientes con PKAN clásico, cursando con nictalopia, pérdida progresiva del campo visual periférico y eventual ceguera (1, 7).
Las alteraciones oculomotoras incluyen alteración de la mirada sacádica, parálisis supranuclear de la mirada, alteración de la convergencia, respuesta optokinética vertical anormal e incapacidad de suprimir reflejo vestíbulo-ocular (1, 7).
Los pacientes sufren episodios de deterioro, intercalados con periodos de estabilidad (1). La esperanza de vida es variable y pueden llegar hasta la edad adulta (1).
PKAN atípica. Sus características clínicas son variadas. Tiene inicio en las tres primeras décadas de la vida (media de 13,6 años). El primer signo puede ser temblor distónico o distonía focal (6). Los defectos del habla incluyen palilalia, taquilalia/taquilogia y disartria (1). La retinopatía es rara en la enfermedad atípica (1, 7). Se presentan trastornos conductuales y del sueño REM, sahos y movimientos periódicos de extremidades (7). Los síntomas psiquiátricos incluyen cambios en la personalidad con impulsividad, estallidos de violencia, depresión y labilidad emocional. Pueden presentar tics motores, verbales, comportamiento obsesivo-compulsivo y, en raras ocasiones, síntomas psicóticos (1, 7). El deterioro cognitivo puede ser parte del fenotipo PKAN de inicio tardío (10).
La afección motora suele ser tardía, con espasticidad, hiperreflexia y otros signos de compromiso del tracto corticoespinal que imitan la deambulación. Puede simular la enfermedad de Parkinson, ya que es posible observar "congelación" durante la deambulación (al girar esquinas o al encontrar variaciones de la superficie) (11), y temblor esencial (12).
Síndrome de HARP: hipoprebetalipoproteinemia, acantocitosis, retinitis pigmentosa y degeneración palidal. Es considerado parte del espectro de la enfermedad (13, 14). Se supone este trastorno es alélico para PKAN (15).
Correlación genotipo-fenotipo
No se ha observado correlación genotipo-fenotipo para PKAN; los individuos con dos mutaciones nulas, que predicen que no hay producción de proteínas, presentan PKAN clásico. Otras combinaciones de mutaciones producen fenotipos clásicos o atípicos sin un patrón predecible (1).
Homocigotos para la mutación missense p.Gly521Arg presentan un fenotipo clásico. Sin embargo, el fenotipo asociado con la condición homocigótica de otros alelos comunes es impredecible. Dos tercios de las personas con PKAN son heterocigotos compuestos, con enfermedad de curso clínico impredecible (1).
Hallazgos patológicos
La neuropatológica se ve limitada por la heterogeneidad de las afecciones que pertenecen a este diagnóstico (16).
En estudios de PKAN en las regiones de la acumulación de hierro se observan cuerpos esferoides mas pequeños respecto a los hallados en otras patologías con acumulación de hierro; estos representan axones inflamados y no se limitan a aquellas porciones del cerebro en las que el hierro se acumula, sino que se han observado en el sistema palidonigral, sustancia blanca y gris del cerebro. Estas lesiones son positivas para ubiquitina en la inmunohistoquímica (16).
Existen otras estructuras esferoidales pequeñas (raras) detectadas por inmunorreactividad para la proteína precursora de amiloide y demostraron menos tinción con inmunohistoquímica antiubiquitina (1, 17, 18).
Neuroimágenes
La RM cerebral es sensible a la presencia y concentración de hierro no hemo en el cerebro, siendo de elección para la investigación y diagnóstico diferencial de los síndromes NBIA (7).
Los casos típicos como atípicos y fenotipo HARP pueden presentar en la fase presintomática el signo de "ojos de tigre". En la secuencia ponderada en T2 se evidencia hiperintensidad en el globo pálido por la vacuolización y gliosis del tejido, rodeado de halo hipointenso por depósito de hierro hallado en las autopsias, con alta correlación con la mutación PANK2. Este hallazgo puede estar ausente en estadios tempranos de la enfermedad (1, 7, 19).
La hiperintensidad palidal central puede desvanecerse a medida que progresa la enfermedad; la identificación de este signo debería impulsar un alto índice de sospecha de PKAN. Los casos de otras neuroferritinopatías con presuntos "ojos de tigre" suelen ser atípicos en apariencia, una cuidadosa evaluación revela contorno irregular y/o desplazamiento lateral de la hiperintensidad central (1, 6, 20).
PKAN debe ser considerado en pacientes con calcificaciones idiopáticas de los ganglios basales, especialmente cuando se limitan al globo pálido. Se han encontrado pacientes con mutaciones del gen PANK2 sin cambios en los genes asociados con calcificaciones idiopáticas de los ganglios basales o distonía que justifiquen los hallazgos y enfermedad de base (21-23).
La neurodegeneración de la membrana mitocondrial asociada a la proteína (MPAN), es una forma diferente de NBIA. Los hallazgos en la neuroimagen puede ser similares a los de PKAN (21).
El signo de "ojos de tigre" no es patognomónico del PKAN, y se ha observado que los adultos pueden presentar el signo en las neuroimágenes con síntomas de enfermedad neurológica, sin hallazgos de mutación en el gen. Puede ser debido a combinación del proceso de envejecimiento asociado a otras condiciones patológicas que pueden dar lugar a este signo en adultos, sugiriéndose que un signo de "ojos de tigre" no puede interpretarse de manera aislada y podrían ser necesarios estudios moleculares antes de establecer el diagnóstico de PKAN, especialmente en adultos (22).
Estudio molecular
El PANK2 es el único gen en el que la mutación es reconocida (1). El análisis secuencial del gen es recomendado después de evidenciar los hallazgos en la RM cerebral. Con la mutación en la secuenciación se confirma el diagnóstico (1).
En un 3-5% de los pacientes no se evidencia alteración genética en el estudio de secuenciación del gen y se debe a deleciones parciales o completas. Si se sospecha clínicamente la enfermedad y no se encuentran mutaciones en la secuenciación o solo se identifica una mutación heterocigota, el análisis de deleción/duplicación del gen debe ser considerado (1).
Diagnóstico diferencial
Algunos pacientes con PKAN se diagnostican como parálisis cerebral o, con menos frecuencia, con enfermedad de Parkinson juvenil. En el diagnóstico diferencial hay que considerar lipofuscinosis ceroide neuronal juvenil, variantes de distonías respondedoras a dopa y enfermedad de cuerpos de Lafora (15). Los errores innatos del metabolismo y trastornos degenerativos con cambios en los núcleos subcorticales, deben tenerse en cuenta (15).
El diagnóstico diferencial se establece con enfermedades con neurodegeneración por acumulación de hierro cerebral o NBIA, que se clasifican como uno de los siguientes: NBIA de aparición temprana, lentamente progresiva, con inicio durante la primera década, lo que incluye la proteína de membrana mitocondrial, asociados a neurodegeneración (MPAN), ácidos grasos, hidroxilasa asociada a neurodegeneración (FAHN) y proteína beta hélice asociada a neurodegeneración (BPAN) (1).
El diagnóstico diferencial de la NBAI de inicio tardío (después de la primera década, con progresión lenta), incluye: PKAN atípico, neuroferritinopatía, aceruloplasminemia, distrofia neuroaxonal atípica, síndrome Kufor-Rakeb, síndrome de Woodhouse-Sakati y NBIA idiopáticas (1).
PKAN puede distinguirse de otras formas de NBIA por los siguientes hallazgos:
En la RM cerebral, en la mayoría de los individuos con NBIA no PKAN NBIA el globo pálido es hipointenso de manera uniforme en las imágenes potenciadas en T2, indicando alto contenido de hierro (17). Este hallazgo es distinto al signo de "ojos de tigre" visto en PKAN. En MPAN las lesiones hiperintensas de la lámina medular medial puede parecerse al signo de "ojos de tigre" (21). El depósito de hierro en el núcleo rojo y núcleo dentado en conjunción con atrofia cerebelosa son comunes en el grupo de NBIA (1).
Las convulsiones están ausentes en PKAN y presentes en las NBIA no PKAN (1). Otros cuatro trastornos pueden mostrar cambios clínicos iniciales similares a los observados en PKAN clásico: discapacidad intelectual ligada al cromosoma X con malformación de Dandy-Walker, alfafucosidosis, síndrome de Leigh (señal hiperintensa simétrica en el globo pálido en la RM ponderada en T2 puede parecerse al signo de PKAN sin hipointensidad circundante, causado por acumulación de hierro) y distrofia neuroaxonal infantil (INAD) (1).
En esta última entidad, en una NBIA de aparición temprana y rápidamente progresiva, con inicio en la primera década, siempre debe pensarse en PKAN y en distrofia neuroaxonal infantil (1, 15). Simonati et al. sugieren tener en consideración esta enfermedad como diagnóstico diferencial (15). Una parte de los individuos muestran señal hipointensa en el globo pálido y sustancia negra, pero el signo de "ojos de tigre" está ausente y la atrofia cerebelosa es común. En INAD esferoides axonales están presentes en el sistema nervioso periférico y en PKAN se encuentran en el SNC (1).
En el diagnóstico diferencial para PKAN adolescente y adulto se incluyen enfermedad de Parkinson juvenil, calcificación cerebral primaria familiar, aceruloplasminemia, neuroferritinopatía, síndrome de Steele-Richardson-Olzewski (parálisis supranuclear progresiva) y enfermedades psiquiátricas primarias (1). Otros trastornos son las ataxias hereditarias de inicio en la infancia (SCA3 y SCA7), distonías como DYT1, enfermedad de Huntington juvenil, corea–acantocitosis, síndrome de LeschNyhan, enfermedad de Wilson, paraplejia espástica hereditaria recesiva, síndrome de Tourette y neuroacantocitosis (1).
Manejo
Se recomienda establecer el grado de la enfermedad, seguimiento y manejo interdisciplinario con apoyo entre neurología, genética, pediatría, fisiatría, ortopedia, fonoaudiología, nutrición y oftalmología pediátrica. Evaluación oftalmológica para determinar la presencia de retinopatía y atrofia óptica. Valoración por fisiatría para determinar el plan de rehabilitación y ayudas de adaptación y dispositivos de movilidad y comunicación según el estado funcional motor y comunicación (1).
Valoración nutricional y seguimiento a largo plazo para determinar de manera oportuna alteraciones nutricionales y necesidad de alimentación por gastrostomía. Valoración por genética ayuda a orientar el estudio genético y asesoría a la familia. Seguimiento por pediatría, ya que estos pacientes tienen mayor riesgo de fracturas sin trauma aparente, infecciones y sangrados de tracto gastrointestinal (1).
Los tratamientos farmacológico y quirúrgico se han centrado en la paliación de los síntomas, ya que no se ha hallado un tratamiento efectivo de la enfermedad (1, 4). El tratamiento sintomático se dirige a la distonía (1).
El manejo puede incluir toxina botulínica intramuscular, palidotomía o talamotomía ablativas, trihexifenidilo y baclofeno oral o intratecal, sobre los que se requieren más estudios para determinar dosis óptima y eficacia. En investigación se halla el uso de baclofeno intraventricular, con el que se ha evidenciado mejoría en la distonía facial y de parte superior del cuerpo (1).
Las personas afectadas con mordedura recurrente de la lengua por distonía orobucolingual severa a menudo requieren de extracción dental como única intervención eficaz (1).
La estimulación cerebral profunda está siendo cada vez más utilizada, con resultados prometedores en PKAN y en otras NBIA (23, 24), evidenciándose en un estudio multicéntrico que los pacientes con distonía severa son los que más se benefician (25).
Los agentes quelantes del hierro se han intentado sin un beneficio claro. La diferiprona, un agente quelante en investigación, a diferencia de otros agentes, atraviesa la barrera hematoencefálica. En estudios en pacientes con PKAN se ha visto reducción estadísticamente significativa de hierro en el globo pálido en la RM cerebral, pero sin cambios en el estado clínico (26).
La existencia de actividad enzimática residual en algunos individuos con PKAN plantea la posibilidad de tratamiento con pantotenato de alta dosis, el cual es el sustrato de la enzima PANK2, pero la eficacia de estos suplementos se desconoce (1).
El ácido docosahexaenóico (DHA) puede desempeñar un papel en la prevención de la degeneración retiniana, aunque aún no se han realizado estudios, proporcionándose como suplemento nutricional oral en forma de ácidos grasos omega-3 grasas (1).
Las terapias que pueden tener una función en otras formas de NBIA no tienen efecto en personas con PKAN e incluyen levodopa/carbidopa y bromocriptina (1).
Conclusiones
Se presentó un paciente de 5 años con un cuadro que cumple los criterios diagnósticos PKAN. Presentamos el caso para que esta entidad se considere en los pacientes que cursen con patología neurodegenerativa con alteración motora de predominio extrapiramidal y principalmente cuando en la neuroimagen se halla depósito de hierro en el globo pálido con el signo de "ojos de tigre" muy sugestivo de la enfermedad, contándose con que se pueden presentar imágenes atípicas y que este signo no es patonomónico de la enfermedad. Por ello, el estudio molecular es necesario.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Referencias
1. GREGORY A, HAYFLICK S. GeneReviews [Internet]; 2002 [actualizado enero 31 de 2013; citado el 12 junio de 2014]. Disponible en http://www. ncbi.nlm.nih.gov/books/NBK1490/. [ Links ]
2. GREGORY A, POLSTER J, HAYFLICK S. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J. Med. Genet. 2009;46:73-80. [ Links ]
3. HAYFLICK S, WESTAWAY S, LEVINSON B, ZHOU B, JOHNSON M, CHING K, GITSCHIER J. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003;348:33-40. [ Links ]
4. DUSEK P, JANKOVIC J, LE W. Iron dysregulation in movement disorders. Neurobiology of Disease 2012;46:1-18. [ Links ]
5. OMIN.ORG [Internet]. EE. UU: OMIM; 1996 [actualizado febrero de 2014; citado el 12 junio de 2014]. Disponible en http://www.omim.org/entry/234200. [ Links ]
6. KRUER M, BODDAERT N. Neurodegeneration with Brain Iron Accumulation: A Diagnostic Algorithm. Semin Pediatr Neurol. 2012;19:67-74. [ Links ]
7. SCHIPPER H. Neurodegeneration with brain iron accumulation. Clinical syndromes and neuroimaging. Biochimica et Biophysica Acta 2012; 1822:350-60. [ Links ]
8. BEKIESINSKA-FIGATOWSKA M, MIERZEWSKA M, JURKIEWICZ E. Basal ganglia lesions in children and adults. Eur J of Radiology 2013;82:837-849. [ Links ]
9. KUSVURAN E, IRMAK S, ET AL. Comparison of several advanced oxidation processes for the decolorization of Reactive Red 120 azo dye in aqueous solution. Journal of Hazardous Materials 2004;109:85- 93. [ Links ]
10. FREEMAN K, GREGORY A, TURNER A, ET AL. Intellectual and adaptive behavior functioning in pantothenate kinase-associated eurodegeneration. J Intellect Disabil Res. 2007;51:417-26. [ Links ]
11. GUIMARAES J, SANTOS J. Generalized freezing in Hallervorden-Spatz syndrome: case report. Eur J Neurol. 1999;6:509-13. [ Links ]
12. YAMASHITA S, MAEDA Y, OHMORI H, UCHIDA Y, HIRANO T, ET AL. Pantothenate kinase-associated neurodegeneration initially presenting as postural tremor alone in a Japanese family with homozygous N245S substitutions in the pantothenate kinase gene. J Neurol Sci. 2004;225:129-33. [ Links ]
13. OMIN.ORG [Internet]. EE. UU.: OMIM; 1996 [actualizado abril de 2010; citado el 12 junio de 2014]. Disponible en http://www.omim.org/entry/607236. [ Links ]
14. HOULDEN H, LINCOLN S, FARRER M, ET AL. Compound heterozygous PANK2 mutations confirm HARP and Hallervorden-Spatz syndromes are allelic. Neurology 2003;61:1423-6. [ Links ]
15. KAZEK B, JAMROZ E, ET AL. Novel PANK2 Gene Mutation: Clinical and Molecular Characteristics of Patients-Short Communication. J Child Neurol. 2007;22:1256-9. [ Links ]
16. SWAIMAN KF. Hallervorden-Spatz syndrome. Pediatr Neurol. 2001;25:102-8. [ Links ]
17. SCHNEIDER S. Syndromes of Neurodegeneration with Brain Iron Accumulation. Semin Pediatr Neurol. 2012;19:57-66. [ Links ]
18. KRUER M, HIKEN M, GREGORY A, ET AL. Novel histopathologic findings in molecularly-confirmed pantothenate kinase-associated neurodegeneration. Brain 2011;134:947-58. [ Links ]
19. HOGARTH P, GREGORY A. New form of neurodegeneration with brain iron accumulation: features associated with MPAN. Neurology 2013;80:268-75. [ Links ]
20. HAYFLICK S. Neurodegeneration with Brain Iron Accumulation: From Genes to Pathogenesis. Semin Pediatr Neurol. 2006;13:182-5. [ Links ]
21. HOGARTH P, GREGORY A, KRUER M, SANFORD L, ET AL. New form of neurodegeneration with brain iron accumulation: features associated with MPAN. Neurology 2013;80:268-75. [ Links ]
22. KUMAR N, BOES C, BABOVIC-VUKSANOVIC D, BOEVE BF. The 'eye-of-the-tiger' sign is not pathognomonic of the PANK2 mutation. Arch. Neurol. 2006;63:292-3. [ Links ]
23. WU Y, HESS C, SINGHAL N. Idiopathic Basal Ganglia Calcifications: An Atypical Presentation of PKAN. Pediatric Neurology 2013,49:351-4. [ Links ]
24. CASTELNAU P, CIF L, VALENTE EM, ET AL. Pallidal stimulation improves pantothenate kinase-associated neurodegeneration. Ann Neurol. 2005;57:738-41. [ Links ]
25. TIMMERMANN L, VOLKMANN J. Deep brain stimulation for treatment of dystonia and tremor. Nervenarzt 2010;81(6):680-7. [ Links ]
26. A two-arm efficacy and safety study of deferiprone in patients with pantothenate kinase-associated neurodegeneration (PKAN) [Internet]. ClinicalTrials.gov [actualizado abril 24 de 2014; citado el 3 de julio de 2014]. Disponible en http://www.clinicaltrials.gov/ct2/show/study/NCT01741532?term=deferiprone&rank=1#contacts. [ Links ]
