










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.3 Bogotá July/Set. 2015
Revisión
Síndrome de temblor y ataxia asociado a frágil X (FXTAS): revisión de la literatura
Fragile X associated tremor/ataxia syndrome: literature review
Wilmar Saldarriaga Gil (1), Jose Vicente Forero Forero (2), Laura Yuriko González Teshima (2), Randi Hagerman (3)
(1) Ginecólogo-Obstetra, Embriología-Genética. Profesor titular, Morfología, Gineco-Obstetricia, Hospital Universitario del Valle, Universidad del Valle, Cali, Colombia.
(2) Estudiante de pregrado en Medicina y Cirugía, Universidad del Valle, Cali, Colombia.
(3) Directora médica, Instituto MIND, Universidad de California en Davis (UC Davis) Estados Unidos. Profesora distinguida, titular de Cátedra en Investigación en Frágil X, Departamento de Pediatría, Escuela de Medicina en UC Davis.
Recibido: 28/07/14. Aceptado: 21/07/15.
Correspondencia: Wilmar Saldarriaga: wilmar.saldarriaga@correounivallle.edu.co
Resumen
El síndrome de temblor y ataxia asociado al síndrome del cromosoma X frágil (FXTAS) es un desorden neurodegenerativo progresivo (1), de inicio tardío, que ocurre entre los portadores de la premutación del gen FMR1 (Fragile X Mental Retardation 1), el cual está estrechamente asociado con el síndrome del cromosoma X frágil (FXS). El FXTAS se caracteriza por déficits neurológicos que incluyen temblor de intención progresivo, ataxia cerebelosa, parkinsonismo, neuropatía periférica, déficits cognitivos y disfunción autonómica (2-4).
El FXTAS surge como una importante opción diagnóstica en hombres con temblor, alteraciones en la marcha y síntomas neurodegenerativos. En general existe subregistro de esta patología dado que es un síndrome recientemente descrito y falta conocimiento de los profesionales de salud al respecto, los cuales, debido a la similitud de su presentación clínica con otros desórdenes neurológicos, generalmente suelen confundir el diagnóstico (5).
En Colombia no se ha documentado la prevalencia de SXF o de FXTAS. Sin embargo, se ha descrito un corregimiento en el Valle del Cauca que tiene una prevalencia de más de cien veces lo reportado en la literatura de SFX, lo que nos sugiere que en Colombia existe subregistro del SFX y de FXTAS.
Esta revisión tiene por objeto difundir los avances del conocimiento de las manifestaciones clínicas, la neurofisiopatología y las posibilidades de tratamiento de los pacientes con FXTAS, y así aumentar diagnóstico y aportar a mejorar la calidad de vida de los afectados y de sus familias.
Palabras clave: ataxia, FXTAS, leucoencefalopatías síndrome de tremor y ataxia asociado al frágil X, síndrome de frágil X, tremor (DECS).
Summary
Fragile X-associated tremor and ataxia syndrome (FXTAS) is a progressive neurodegenerative disorder (1) of late onset that occurs among the premutation carriers of the FMR1 (Fragile X mental retardation 1) gene; which is also associated with Fragile X Syndrome (FXS). FXTAS is characterized by neurologic deficits that include progressive intention tremor, cerebellar ataxia, parkinsonism, peripheral neuropathy, cognitive deficits in memory and executive function, and autonomic dysfunction.
FXTAS is emerging as an important diagnostic option for men with tremor, gait disorders and neurodegenerative symptoms. In general there is a subdiagnosis of this disease as it is a recently described syndrome and because of the lack of knowledge from health professionals; whom, due to the similarity of their clinical presentation with other neurological disorders, generally tend to confuse the diagnosis.
In Colombia the prevalence of FXS or FXTAS has not been documented. However, a township in Valle del Cauca has a prevalence of more than 100 times the worldwide prevalence of FXS reported in the literature, which suggests that in Colombia there is underreporting of FXS and FXTAS.
This review aims to disseminate the advances in knowledge of the clinical manifestations, the neurophysiopathology and treatment options for patients with FXTAS; and thus increase diagnosis and contribute to improving the quality of life of those affected and their families.
Key words: Ataxias, Fragile X Mental Retardation Syndrome, Fragile X tremor/Ataxia syndrome, Fxtas, Tremors, Leukoencephalopathy (MeSH).
Introducción
El síndrome de temblor y ataxia asociado al frágil X (FXTAS) es un desorden neurodegenerativo progresivo (1), de inicio tardío, que ocurre entre los portadores de la premutación del gen FMR1 (Fragile X Mental Retardation 1). Por premutación se entiende la presencia de una expansión de 55-200 repeticiones de la tripleta CGG en la región 5'UTR de dicho gen, siendo el número normal de repeticiones alrededor de 30. El FXTAS está caracterizado por déficits neurológicos que incluyen temblor de intención progresivo, ataxia cerebelosa, parkinsonismo, neuropatía periférica, déficits cognitivos en memoria y funciones ejecutivas, y disfunción autonómica (2-7).
A diferencia de lo que ocurre en la mayoría de los pacientes con la mutación completa, es decir, con el síndrome de cromosoma X frágil, y en quienes tienen el gen FMR1 completamente metilado ("silenciado"), lo que conduce a un expresión mínima o inexistente del mRNA y un subsecuente déficit de la proteína del FMR1 (FMRP) (8), se ha visto que los pacientes con FXTAS tienen sobreexpresión del gen, lo cual lleva a una aparente toxicidad por exceso de su mRNA (Figura 1). Sin embargo, también se han descrito algunos casos del síndrome en portadores dentro de la zona gris (45-54 repeticiones de CGG) (9, 10) y en individuos con la mutación completa (> 200 repeticiones de CGG) que tienen el gen parcial o completamente desmetilado ("activo") (11, 12).
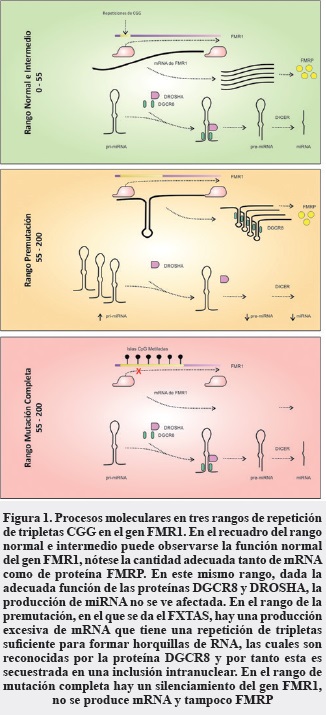
Si bien en el FXS clásico también se presentan alteraciones en el neurodesarrollo, estas no deben ser confundidas con FXTAS, ya que la neurofisiopatología de la enfermedad es muy diferente. Así, a pesar de que las dos entidades son debidas a mutaciones por expansión de tripletas en el gen FMR1, el déficit de FMRP genera características físicas de aparición temprana tales como orejas prominentes, cara alargada, talla en percentiles altos, obesidad y macroorquidismo, acompañadas de discapacidad intelectual y problemas del lenguaje, fenotipo que no se encuentra en FXTAS.
En Colombia no se ha documentado la prevalencia de SXF o de FXTAS. Sin embargo, se ha descrito un corregimiento en el Valle del Cauca que tiene una prevalencia de más de cien veces lo reportado en la literatura de SXF, del cual ha existido migración a personas jóvenes que podrían ser portadoras y en ese momento no haber presentado síntomas o tenido hijos afectados. Además, existe una asociación de padres de pacientes con SXF, lo que nos sugiere que en Colombia hay pacientes con FXTAS que no se han diagnosticado y podrían realizarse estudios para establecer prevalencias del SFX y de FXTAS (7).
Esta revisión desarrolla en profundidad epidemiología, criterios diagnósticos clínicos e imagenológicos, neuro-fisio-patología, bases genéticas y tratamiento del síndrome de temblor y ataxia asociado al X frágil.
Epidemiología
La prevalencia del alelo mutado de FXTAS es común entre la población general y varía según el grupo poblacional que se esté evaluando (1, 2). En España se encontró una prevalencia de 1 en 130 mujeres y 1 en 250 hombres (13). En Israel la prevalencia en mujeres fue similar con datos de 1 en 113-157 mujeres (14). Seltzer et al. , en 2012, presentaron estudios realizados en una población estadounidense, en los que encontraron la premutación del gen FMR1 con una frecuencia de 1 en 151 mujeres y 1 en 468 hombres de la población general, es decir, a una razón de prevalencia entre mujeres y hombres de 3.1:1 respectivamente (3). Ya que aproximadamente el 40% de los hombres portadores y del 8-16% de las mujeres portadoras desarrollarán FXTAS a medida que avanza su edad, ello permite estimar una prevalencia aproximada de 1 en 3.000 (15). Esta prevalencia de FXTAS es similar a la de otras enfermedades neurológicas como la atrofia multisistémica, la esclerosis lateral amiotrófica o la parálisis supranuclear progresiva (16). Para los hombres, el FXTAS podría clasificarse como una de las enfermedades neurológicas progresivas de inicio tardío más comunes asociadas a la mutación de un gen único (3).
A pesar de que la prevalencia de la premutación del gen FMR1 es tan elevada, la prevalencia del FXTAS es menor de lo estimado, lo que podría indicar un posible subregistro de esta patología. Esto podría deberse a que el FXTAS presenta unas manifestaciones clínicas similares a otros desórdenes neurológicos, lo que aumenta la probabilidad de hacer diagnóstico equivocado, acompañado de la poca remisión a neurología de estos pacientes (5).
La penetrancia promedio de FXTAS entre los portadores de la premutación a los 50 años es del 40% en hombres (2, 17) y del 8% en mujeres (15, 18), es decir que no se debe asumir que todos los portadores de la premutación van a desarrollar este síndrome. Sin embargo, el porcentaje de penetrancia va a variar según la edad del portador, el cual incrementa su riesgo de desarrollar FXTAS a medida que aumenta su edad. Jacquemont et al. estudiaron las familias conocidas en California y encontraron que los signos de temblor y ataxia ocurrían en 17%, 38%, 47% y 75% de hombres portadores en edades entre los 50-59, 60-69, 70-79, y mayores de 80 años, respectivamente (19).
Posterior a la descripción de FXTAS por primera vez en el 2001 (20), se empezaron a identificar los pacientes portadores de la premutación entre las poblaciones clínicas de diversos centros de investigación. Se realizaron tamizajes de los bancos de DNA disponibles y se lograron identificar coincidencias entre los pacientes diagnosticados con ataxia y parkinsonismo, los cuales resultaron ser portadores de la premutación para el gen FMR1 (5). El 2,2% de los pacientes con ataxia de inicio en la adultez, el 4% de las pacientes con ataxia y un examen genético negativo para ataxia espinocerebelar autosómica dominante (SCA) y el 5% de los pacientes hombres con ataxia y prueba negativa para SCA, presentaron premutación en el gen FMR1 (5). El FXTAS es entonces el diagnóstico adecuado para aquellos que tengan la premutación y padezcan estos síntomas neurológicos. La posible relación de este hallazgo con otras condiciones neurodegenerativas es desconocida, pero también se ha visto que pueden coexistir. Así, en pacientes con parkinsonismo y atrofia multisistémica se identificó la premutación entre el 0-4% de los pacientes, y en 0-1% de los pacientes con enfermedad de Parkinson (5).
Manifestaciones clínicas y diagnóstico de FXTAS
Las manifestaciones clínicas tienen un inicio típico alrededor de los 60-65 años de edad. El FXTAS se caracteriza por la presentación de temblor de intención y ataxia cerebelosa, que son los dos criterios clínicos mayores para su diagnóstico (1, 2, 5, 16, 21). También, y aunque estos síntomas motores son usualmente encontrados, en ocasiones el FXTAS se presenta con detrimento cognitivo aislado, que puede incluir posteriormente temblor y/o ataxia. FXTAS puede ocurrir junto con enfermedad de Parkinson, patología con la cual comparte características clínicas como temblor, ataxia, bradiquinesia, ansiedad, alteraciones del ánimo y cognitivas. No obstante, la ataxia típica de Parkinson no corresponde a la ataxia cerebelosa característica del FXTAS (4).
Además de estos, existen otros criterios diagnósticos menores que incluyen problemas de memoria a largo plazo de intensidad moderada-severa, asociado a disminución en el volumen del hipocampo, hallazgo que se ha reportado tanto en hombres como en mujeres (15); déficit en las funciones ejecutivas, alteraciones psiquiátricas (ansiedad, depresión, demencia, irritabilidad, pocas habilidades sociales) (18) y parkinsonismo (1, 17). Cabe resaltar que, a diferencia del FXS, estos pacientes no presentan retardo mental, tienen un IQ normal y en la mayoría de los casos preservan la comprensión verbal y el lenguaje a pesar de los síntomas motores, la demencia y la disfunción ejecutiva prominente que lleva a déficit en la memoria operativa y de aprendizaje, la secuenciación temporal, la velocidad de procesamiento de la información, entre otras, alterando significativamente la calidad de vida e independencia de estas personas (16).
Seritan et al. (5) demostraron que 42% de los pacientes masculinos que desarrollaron FXTAS tardíamente desarrollaron un déficit en el desarrollo cognitivo significativo y cumplieron con los criterios diagnósticos para demencia. Igualmente se identificaron, a través de patrones electroclínicos, tres tipos de temblores predominantes: 35% de los pacientes presentaron temblor esencial con temblor de pequeña amplitud, 29% temblor cerebeloso bilateral próximo distal y el 12% de temblor tipo parkinsonismo unilateral asociado al reposo (5).
Sin embargo, las manifestaciones clínicas por sí solas no permiten realizar un diagnóstico certero de FXTAS, el cual debe contar con la presencia de ciertos hallazgos radiológicos específicos, tales como la hiperintensidad del pedúnculo cerebelar medio (MCP por las siglas en inglés de Medium Cerebellar Peduncle) observada en imagen de resonancia magnética de cerebro (Figura 2A), aunque este no es patognomónico. Pese a ello, constituye un criterio radiológico mayor (5) que debe ir acompañado de los criterios clínicos no radiológicos mencionados para constituir un diagnóstico. El signo de MCP solo se encuentra en el 60% de los hombres y el 13% de las mujeres con FXTAS; estos pacientes presentan un déficit cognitivo más severo y una historia más larga de síntomas de degeneración neurológica que los individuos con FXTAS sin él.
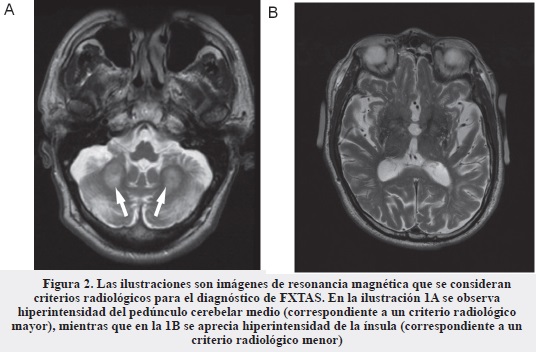
Entre otros hallazgos neurológicos se incluyen criterios radiológicos menores, también identificados a través de RMN T2, como la hiperintensidad de la materia blanca en diferentes zonas del sistema nervioso central como los núcleos pontinos, la ínsula (Figura 2B), el esplenio del cuerpo calloso y la región periventricular. Por otro lado, el segundo criterio radiológico menor sería la atrofia cerebral severa generalizada (1, 5, 18). La atrofia se hace más evidente en la parte frontal de la corteza, incluyendo las áreas dorsomediana y dorsolateral de la corteza prefrontal (1, 16).
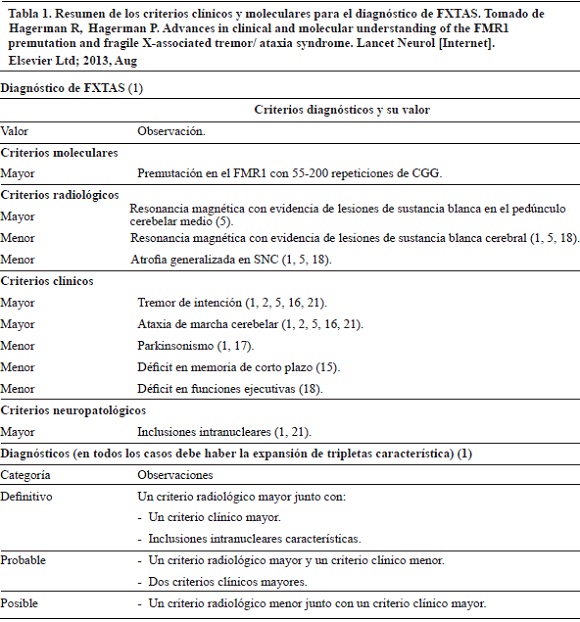
Según Hagerman et al. (1, 21), para un diagnóstico definitivo de FXTAS (Tabla 1) se necesita la presencia de un criterio clínico y radiológico mayor, o un criterio radiológico mayor y la presencia de inclusiones intranucleares redondeadas típicas de FXTAS en neuronas y astrocitos (1, 21). Estas inclusiones tienen una distribución amplia a través de todo el cerebro y el tallo cerebral (1, 17, 21). En el caso de un diagnóstico probable, se debe presentar un criterio radiológico mayor acompañado de un criterio clínico menor, o por el contrario, dos criterios clínicos mayores, es decir, la presentación conjunta de temblor de intención y ataxia cerebelar que característicamente se pueden identificar en estos pacientes al examen clínico. Por último, un diagnóstico posible de FXTAS se constituye a partir de la presencia de un criterio radiológico menor y un criterio clínico mayor (1, 16). De manera que el diagnóstico de este síndrome se hace mucho más complicado que la observación de temblor y ataxia en pacientes con la premutación del gen FMR1 identificada (1, 2, 4).
Neuropatología
Hagerman et al. (22-24) han realizado varias observaciones acerca de la neuropatología del FXTAS a partir de diecinueve tejidos del sistema nervioso central extraídos post mortem de dieciocho casos con FXTAS y un paciente portador de la premutación pero sin las manifestaciones clínicas del síndrome. El primer hallazgo que resulta de importancia es la presencia de inclusiones exclusivamente intranucleares que pueden ser observadas con tinción de hematoxilina y eosina o con inmunohistoquímica dirigida a ubiquitina. Estas inclusiones son esféricas, solitarias y presentes en neuronas y astrocitos (ausentes en oligodendroglia) distribuidas en el sistema nervioso central y periférico, e incluso presentes en diferentes órganos como testículos y glándula suprarrenal (22-25).
Es importante mencionar que existe una proporción directa lineal entre el número de repeticiones de la tripleta CGG y el número de inclusiones presentes en el tejido. Además, en las mujeres tiende a formarse un menor número de estas inclusiones al compararlas con los hombres. Se presume que esto se debe a la influencia del segundo cromosoma X sin la mutación y la tasa de activación del mismo (26).
En los estudios post mortem se observó una severa disminución de la sustancia blanca cerebral y cerebelar que es consistente con los hallazgos por RMN. Respecto de lo anterior, el compromiso del pedúnculo cerebelar medio (Figura 2A) es detectado en un 60% de los pacientes por estudios imagenológicos, pero histopatológicamente se encuentra en siete de cada ocho casos brindando una noción de la sensibilidad de la RMN en el diagnóstico de esta patología (24, 26).
A pesar de que la atrofia cerebral, el adelgazamiento del cuerpo calloso y la depleción de células de Purkinje son hallazgos prominentes en la neuropatología del FXTAS, es importante resaltar que hay pocas inclusiones en los núcleos pontinos presentes en la región basilar del puente y casi ninguna en el núcleo de las células de Purkinje. Es posible que las células de Purkinje con inclusiones hayan sufrido una muerte celular más temprana o, tal vez, no puedan formar inclusiones y esto las haga más vulnerables a la misma (26).
Bases genéticas y moleculares
Es claro que el mRNA elevado del gen FMR1 cumple un papel importante en la patogenia del FXTAS. Existe cierta deficiencia de proteína FMRP entre los individuos premutados y los que tienen un alelo en el rango normal, pero no es una diferencia significativa (26-29). La premutación, sin embargo, puede, mediante de la toxicidad por RNA, causar a las neuronas una vulnerabilidad a muerte celular temprana (30) debida a golpes genéticos secundarios, como: (31) toxicidad ambiental (32), los efectos deletéreos de las convulsiones recurrentes (33) o incluso niveles disminuidos de FMRP que se han asociado a autismo y/o TDAH en algunos portadores de la premutación (34). Aunque el papel patogénico del mRNA puede estar restringido a los eventos desencadenantes en el neurodesarrollo del paciente, son necesarias distintas injurias para que este síndrome neurodegenerativo se consolide (26). Es posible que estas sean causadas por alcoholismo crónico, abuso de opioides, tabaquismo, hipertensión no tratada, depresión, hipotiroidismo o deficiencias vitamínicas, y exposición a anestesia prolongada durante cirugía.
Se han propuesto diferentes modelos de toxicidad por el exceso de mRNA y entre ellos el de mayor evidencia es el del secuestro de proteínas ligadoras de RNA, el cual tiene como modelo referente la distrofia miotónica (DM) (1, 35, 36). Según este modelo, el mRNA en exceso de FMR1 que contiene la expansión de la tripleta CGG, forma horquillas que son reconocidas por proteínas ligadoras de RNA muy importantes para la función de neuronas y astrocitos. La combinación de estas proteínas secuestradas y el exceso de mRNA lleva a la formación de inclusiones en los tipos celulares mencionados (26). Las proteínas que se unen, en modelos in vitro, a estas estructuras en horquilla, son: hnRNP A2/B1 (26, 35, 37), Purα (26, 38, 39), Sam68 (26, 40) y DGCR8 (Pasha) (26, 41, 42), entre otras con menor evidencia en modelos murinos (42).
Sellier et al. (42) demostraron el papel central de una de estas proteínas en la neurodegeneración presente en FXTAS. Dicha proteína es DGCR8, la cual, junto a la proteína Drosha, lleva a cabo el primer paso de la maduración de los miRNA (Figura 1). Las cuatro observaciones principales que exponen en su investigación son:
-
DGCR8 se une preferencialmente a RNA con más de cincuenta y cinco repeticiones de la tripleta CGG, lo que coincide con el límite inferior del rango de la premutación. Esta situación no ocurre en las otras proteínas mencionadas, siendo una condición importante para explicar el por qué se presenta la patología casi exclusivamente en este rango de premutación.
-
Los niveles de miRNA en células cultivadas con la premutación y en tejido del sistema nervioso central de pacientes con FXTAS se encuentran disminuidos, mientras que los niveles de miRNA primarios (pri-miRNA) se encuentran aumentados. Esto se debe a que el complejo Drosha-DGCR8 escinde una parte del pri-miRNA para convertirse en pre-miRNA y en el citoplasma ser convertido a miRNA. Al no haber DGCR8 funcional disponible en núcleo celular, se acumulará sustrato (pri-miRNA) y faltará producto (miRNA) (43, 44).
-
La sobreexpresión de DGCR8 aumentó la viabilidad neuronal y la complejidad dendrítica en modelos con repeticiones CGG expandidas.
-
La sobreexpresión de Drosha no tiene ningún efecto sobre los dos aspectos mencionados en el anterior numeral. Esto indica que es la disfunción de DGCR8 y no de Drosha la que causa los hallazgos expuestos en el numeral 2.
La insuficiencia funcional de esta proteína llevará entonces a una desregulación génica debido a la deficiencia del mecanismo silenciador postranscripcional de los miRNA. Esta desregulación puede resultar en diversos mecanismos patogénicos que no han sido especificados aún; sin embargo, este modelo de toxicidad puede no ser el único presente en la patología. Otros modelos han sido postulados como posibles mediadores. Entre ellos, los más importantes son:
-
Interacción entre el mRNA y proteínas con dominios similares a priones que llevan a una activación del dominio priongénico y a la activación de una cascada de agregación proteica similar a la formación de placas amiloides en Alzheimer (45). De hecho la proteína hnRNP A2/B1 ha sido clasificada como altamente priongénica según King et al. (46). Sin embargo, casi todas las agregaciones proteicas formadas por priones son citoplasmáticas, mientras que las inclusiones observadas en FXTAS son intranucleares (26).
-
Traducción del mRNA con repetición de tripletas expandidas mediante un proceso denominado traducción RAN, traducción no dependiente del codón de inicio AUG asociada a repetición de tripletas. Este proceso conduce a la producción de proteínas de poliglicina que son tóxicas para la neurona. (47). Es posible que estos mecanismos ocurran de manera sobreagregada en FXTAS.
Tratamiento
Actualmente no existe un tratamiento específico para FXTAS (1, 2). El manejo se da principalmente para cada uno de los síntomas que presentan los pacientes, basándose en diferentes estrategias de intervención y monitoreo en búsqueda de mejorías de las manifestaciones clínicas. Básicamente se trabajan cinco líneas de acción dentro del manejo de estos pacientes, las cuales buscan, por un lado, aliviar los síntomas neurológicos y psiquiátricos, al igual que desarrollar y mantener un monitoreo constante y detallado de la progresión y degeneración de habilidades motoras y cognitivas. Es ideal que el paciente reciba una atención interdisciplinaria, permitiendo fortalecer y propulsar la evolución del individuo de forma positiva, al contar con diferentes especialistas en desórdenes del movimiento, del habla, de funciones ejecutivas y motoras, al igual que un acompañamiento constante con terapia ocupacional y fisioterapia que le permitan recuperar su funcionalidad e independencia (2). Por último, es de vital importancia realizar consejería genética para el paciente y su familia, facilitando la comprensión de este proceso y generando conciencia sobre el riesgo que existe para todos los familiares de sufrir este tipo de síndromes (2, 5).
El monitoreo de los pacientes debe ser constante debido a que es una enfermedad neurodegenerativa crónica. Berry et al. (48) estimaron que el promedio de inicio de los temblores y la ataxia ocurre a los 60 años, a partir de los cuales se describe un proceso de deterioro neurológico y motor; se evidenció que en los dos años siguientes al primer síntoma motor se presenta la ataxia (48), seguida a los seis años por aumento en la frecuencia de caídas; a los quince años la necesidad de aparatos que permitan la movilidad; finalmente, a los veintiún años del inicio de los síntomas se espera que el paciente muera en condiciones de discapacidad absoluta, postrado en cama, disartrósico, disfágico, con ausencia total de control de esfínteres y parkinsonismo severo. De manera que la expectativa de vida entre las personas que desarrollan este síndrome es de 5-25 años, dependiendo de las condiciones de cada caso (48).
Diferentes estudios han reportado el uso positivo de fármacos como betabloqueadores, primidona o topiramato para el control del temblor en FXTAS, siendo los bloqueadores betaadrenérgicos como el propanolol los medicamentos más efectivos para el tratamiento del temblor esencial típico de este síndrome. (2, 49). En caso de que los temblores no cedan, la segunda línea de tratamiento incluye topiramato y anticonvulsivantes (49). Igualmente, se han utilizado terapias como estimulación cerebral profunda (DBS) para el temblor de intención severo (1), pero este procedimiento solo se debe realizar cuando el temblor representa el principal problema, ya que la ataxia podría empeorar con la DBS, aunque en ocasiones puede mejorar. La toxina botulínica puede también mejorar el temblor severo en estos pacientes (49).
En cuanto a la ataxia, fármacos como la amantidina pueden llegar a ser beneficiosos en algunos casos (50, 51). Se realizó un estudio controlado de memantina en FXTAS que fracasó para el tratamiento de temblor y ataxia, aunque se pueden ver mejorías en ciertos pacientes individualizando la administracion de este medicamento que bloquea la toxicidad del glutamato (52, 53). A pesar de que la terapia farmacológica ha demostrado tener efectos positivos, esta necesita del apoyo de la terapia física como herramienta para el mejoramiento de la fuerza y estabilidad de los pacientes con ataxia, especialmente a medida que estos envejecen. De todas maneras, se necesitan estudios controlados que puedan afirmar estas relaciones.
Los pacientes con FXTAS deben ser manejados en forma íntegra e idealmente por un grupo interdisciplinario que incluya neurólogo, genetista, terapistas, psiquiatra, entre otros. La atención requiere incluir el manejo de los síntomas neurológicos, pero también se deben intervenir la ansiedad y depresión para mejorar la actitud de los pacientes y sus familias. Con este fin se han indicado inhibidores de la recaptación de serotonina (IRSS) y benzodiacepinas, las cuales, a pesar de poder ser útiles, causan sedación, lo que puede llegar a ser inconveniente para muchos pacientes (1, 5, 49). Tanto los IRSS como el ejercicio aumentan la neurogénesis, lo cual puede ser benéfico en FXTAS (1). Asimismo, el uso de folato y vitamina B12 se ha recomendado por su efecto en la disminución de la tasa de atrofia cerebral en la población general al disminuir los niveles de homocisteína (1).
Actualmente se están investigando diversos tratamientos derivados del entendimiento o de las teorías de la fisiopatología del FXTAS que, a pesar de ser prometedores, necesitan de mayores estudios. Se trabaja con agonistas GABA debido al déficit de GABAA que presentan los pacientes de FXTAS, restaurando un poco el desbalance inhibitorio y excitatorio que se cree existe en este síndrome. (1, 54).
Conclusión
El FXTAS surge como una importante opción diagnóstica en pacientes con temblor, ataxia y síntomas neurodegenerativos con hallazgos imagenológicos como disminución en el volumen del hipocampo, hiperintensidad del pedúnculo cerebelar medio, de la materia blanca en los núcleos pontinos, la ínsula, el esplenio del cuerpo calloso y la región periventricular, y como un campo de investigación relevante en Colombia, dado que en nuestro país existe una población en la que se ha reportado la prevalencias más elevada de FXS en la literatura, de donde han migrado gran cantidad de personas al resto de Colombia y el mundo. Existen pruebas moleculares para confirmar el diagnóstico y terapia farmacológica y no farmacológica que mejoran la calidad de vida en los afectados por FXTAS. El diagnóstico trasciende del paciente a sus familiares dada la posibilidad de que algunos sean portadores de la premutación del SFX, de presentar FXTAS, y de que quienes están en edad reproductiva puedan tener hijos con SFX.
Conflicto de intereses.
Los autores declaran no tener conflicto de intereses.
Referencias
1. HAGERMAN R, HAGERMAN P. Advances in clinical and molecular understanding of the FMR1 premutation and fragile X-associated tremor/ataxia syndrome. Lancet Neurol. 2013, Aug;12(8):78698 [citado 2014 marzo 20]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3922535&tool=pmcentrez&rendertype=abstract. [ Links ]
2. BERRY-KRAVIS E, ABRAMS L, COFFEY SM, HALL DA, GRECO C, GANE LW, ET AL. Fragile X-associated tremor/ataxia syndrome: clinical features, genetics, and testing guidelines. Mov Disord [Internet]. 2007, Jul;22(14):2018-30. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17618523. [ Links ]
3. SELTZER MM, BAKER MW, HONG J, MAENNER M, GREENBERG J, MANDEL D. Prevalence of CGG expansions of the FMR1 gene in a US population-based sample. Am J Med Genet B Neuropsychiatr Genet. 2012, Jul;159B(5):589-97 [citado 2014 marzo 20]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3391968&tool=pmcentrez&rendertype=abstract. [ Links ]
4. BOURGEOIS JA, COGSWELL JB, HESSL D, ZHANG L, ONO MY, TASSONE F, ET AL. Cognitive, anxiety and mood disorders in the fragile X-associated tremor/ataxia syndrome. Gen Hosp Psychiatry 2007;29(4):349-56 [citado 2014 abril 16]. Disponible en: http://www.ncbi.nlm.nih.gov/ pubmed/17591512. [ Links ]
5. HALL DA, O'KEEFE JA. Clinical neurogenetics: fragile x-associated tremor/ataxia syndrome. Neurol Clin. 2013, Nov;31(4):1073-84 [citado 2014 abril 15]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24176424. [ Links ]
6. BERRY-KRAVIS E, ABRAMS L, COFFEY SM, HALL DA, GRECO C, GANE LW, ET AL. Fragile X-Associated Tremor / Ataxia Syndrome: Clinical Features, Genetics, and Testing Guidelines 2007;22(14):2018-30. [ Links ]
7. SALDARRIAGA W, TASSONE F, GONZÁLEZ LY, FORERO JV, AYALA S, HAGERMAN R. Fragile X Syndrome [Internet]. Colombia Médica; 2014. p. 190-8. Disponible en: http://colombiamedica.univalle.edu.co/index.php/comedica/article/view/1810/2577 Consultado en abril 9 de 2015. [ Links ]
8. BAGNI C, TASSONE F, NERI G, HAGERMAN R. Science in medicine Fragile X syndrome: causes, diagnosis, mechanisms, and therapeutics 2012;122(12). [ Links ]
9. LIU Y, WINARNI T, ZHANG L, TASSONE F, HAGERMAN R. Fragile X-associated tremor/ataxia syndrome (FXTAS) in grey zone carriers. Clin Genet. 2013;84:74-7. [ Links ]
10. HALL D, TASSONE F, KLEPITSKAYA O, LEEHEY M. Fragile X Associated Tremor ataxia syndrome in FMR1 gray zone allele carriers. Movement Disorders; 2012. p. 297-301. [ Links ]
11. PRETTO DI, HUNSAKER MR, CUNNINGHAM CL, GRECO CM, HAGERMAN RJ, NOCTOR SC, ET AL. Intranuclear inclusions in a fragile X mosaic male. Transl Neurodegener. 2013;2:10 [Internet]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3668897&tool=pmcentrez&rendertype=abstract. [ Links ]
12. LOESCH DZ, SHERWELL S, KINSELLA G, TASSONE F, TAYLOR A, AMOR D, ET AL. Fragile X-associated tremor/ataxia phenotype in a male carrier of unmethylated full mutation in the FMR1 gene. Clin Genet. 2012;82:88-92. [ Links ]
13. FERNÁNDEZ-CARVAJAL I, WALICHIEWICZ P, XIAOSEN X, PAN R, HAGERMAN PJ, TASSONE F. Screening for expanded alleles of the FMR1 gene in blood spots from newborn males in a Spanish population. J Mol Diagn. 2009, Jul;11(4):324-9 [citado 2014 marzo 25]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2710709&tool=pmcentrez&rendertype=abstract. [ Links ]
14. TOLEDANO-ALHADEF H, BASEL-VANAGAITE L, MAGAL N, DAVIDOV B, EHRLICH S, DRASINOVER V, ET AL. Fragile-X carrier screening and the prevalence of premutation and full-mutation carriers in Israel. Am J Hum Genet [Internet]. 2001, Aug;69(2):351-60. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1235307&tool=pmcentrez&rendertype=abstract. [ Links ]
15. PIROZZI F, TABOLACCI E, NERI G. The FRAXopathies: definition, overview, and update. Am J Med Genet A. 2011, Aug;155A(8):1803-16. [citado 2014 abril 16]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/21739597. [ Links ]
16. LEEHEY MA. Fragile X Y Associated Tremor / Ataxia Syndrome: Clinical Phenotype, Diagnosis, and Treatment. 2009;57(8):830-6. [ Links ]
17. WANG JY, HAGERMAN RJ, RIVERA SM. A multimodal imaging analysis of subcortical gray matter in fragile X premutation carriers. Mov Disord. 2013, Aug;28(9):1278-84 [citado 2014 abril 16]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23649693. [ Links ]
18. BOURGEOIS JA, SERITAN AL, CASILLAS EM, HESSL D, SCHNEIDER A, YANG Y, ET AL. Lifetime prevalence of mood and anxiety disorders in fragile X premutation carriers. J Clin Psychiatry 2011, Feb;72(2):175-82 [citado 2014 marzo 20]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/20816038. [ Links ]
19. JACQUEMONT S, HAGERMAN RJ, LEEHEY MA, HALL DA, LEVINE RA, BRUNBERG JA, ET AL. Penetrance of the fragile X-associated tremor/ataxia syndrome in a premutation carrier population. JAMA 2004;291:460-9. [ Links ]
20. HAGERMAN RJ, LEEHEY M, HEINRICHS W, TASSONE F, WILSON R, HILLS J, ET AL. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, Jul;57(1):127-30 [citado 2015 julio 13]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/11445641. [ Links ]
21. HAGERMAN P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 2013, Jul;126(1):1-19 [citado 2014 abril 15]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3904666&tool=pmcentrez&rendertype=abstract. [ Links ]
22. GRECO CM, BERMAN RF, MARTIN RM, TASSONE F, SCHWARTZ PH, CHANG A, ET AL. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS). Brain 2006;129:243-55. [ Links ]
23. GRECO CM, HAGERMAN RJ, TASSONE F, CHUDLEY AE, BIGIO MR DEL, JACQUEMONT S, ET AL. Neuronal intranuclear inclusions in a new cerebellar tremor/ataxia syndrome among fragile X carriers. Brain 2002, Aug;125(Pt 8):1760-71. [Internet]. Disponible en: http://www.ncbi.nlm.nih gov/pubmed/12135967. [ Links ]
24. TASSONE F, GRECO CM, HUNSAKER MR, SERITAN AL, BERMAN RF, GANE LW, ET AL. Neuropathological, clinical and molecular pathology in female fragile X premutation carriers with and without FXTAS. Genes Brain Behav. 2012;11:577-85. [Internet]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22463693. [ Links ]
25. HUNSAKER MR, GRECO CM, SPATH MA, SMITS APT, NAVARRO CS, TASSONE F, ET AL. Widespread non-central nervous system organ pathology in fragile X premutation carriers with fragile X-associated tremor/ataxia syndrome and CGG knock-in mice. Acta Neuropathol. 2011;122:467-79. [ Links ]
26. HAGERMAN P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 2013;126:1-19. [Internet]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3904666&tool=pmcentrez&rendertype=abstract. [ Links ]
27. TASSONE F, HAGERMAN RJ, CHAMBERLAIN WD, HAGERMAN PJ. Transcription of the FMR1 gene in individuals with fragile X syndrome. Am J Med Genet. 2000;97:195-203. [ Links ]
28. TASSONE F, HAGERMAN RJ, LOESCH DZ, LACHIEWICZ A, TAYLOR AK, HAGERMAN PJ. Fragile X males with unmethylated, full mutation trinucleotide repeat expansions have elevated levels of FMR1 messenger RNA. Am J Med Genet. 2000;94:232-6. [ Links ]
29. KENNESON A, ZHANG F, HAGEDORN CH, WARREN ST. Reduced FMRP and increased FMR1 transcription is proportionally associated with CGG repeat number in intermediate-length and premutation carriers. Hum Mol Genet. 2001;10:1449-54. [ Links ]
30. CHEN Y, TASSONE F, BERMAN RF, HAGERMAN PJ, HAGERMAN RJ, WILLEMSEN R, ET AL. Murine hippocampal neurons expressing Fmr1 gene premutations show early developmental deficits and late degeneration. Hum Mol Genet. 2009;19(1):196-208. [ Links ]
31. LOZANO R, SUMMERS S, LOZANO C, MU Y, HESSL D, NGUYEN D, ET AL. Association between macroorchidism and intelligence in FMR1 premutation carriers. Am J Med Genet A. 2014, Jun;1-6 [citado 2014 junio 11]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/24903624. [ Links ]
32. GARCÍA-AROCENA D, HAGERMAN PJ. Advances in understanding the molecular basis of FXTAS. Hum Mol Genet. 2010;19. [ Links ]
33. CHONCHAIYA W, AU J, SCHNEIDER A, HESSL D, HARRIS SW, LAIRD M, ET AL. Increased prevalence of seizures in boys who were probands with the FMR1 premutation and co-morbid autism spectrum disorder. Hum Genet. 2012;131:581-9. [ Links ]
34. FARZIN F, PERRY H, HESSL D, LOESCH D, COHEN J, BACALMAN S, ET AL. Autism spectrum disorders and attention-deficit/hyperactivity disorder in boys with the fragile X premutation. J Dev Behav Pediatr. 2006;27:S137-44. [ Links ]
35. ECHEVERRÍA GV, COOPER TA. RNA-binding proteins in microsatellite expansion disorders: Mediators of RNA toxicity. Brain Research; 2012. p. 100-11. [ Links ]
36. UDD B, KRAHE R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol. 2012;11:891-905. [Internet]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/22995693. [ Links ]
37. SOFOLA OA, JIN P, QIN Y, DUAN R, LIU H, HARO M DE, ET AL. RNA-Binding Proteins hnRNP A2/B1 and CUGBP1 Suppress Fragile X CGG Premutation Repeat-Induced Neurodegeneration in a Drosophila Model of FXTAS. Neuron. 2007;55:565-71. [ Links ]
38. AUMILLER V, GRAEBSCH A, KREMMER E, NIESSING D, FÖRSTEMANN K. Drosophila Pur-α binds to trinucleotide-repeat containing cellular RNAs and translocates to the early oocyte. RNA Biology; 2012. p. 633-43. [ Links ]
39. JIN P, DUAN R, QURASHI A, QIN Y, TIAN D, ROSSER TC, ET AL. Pur α Binds to rCGG Repeats and Modulates Repeat-Mediated Neurodegeneration in a Drosophila Model of Fragile X Tremor/Ataxia Syndrome. Neuron. 2007;55:556-64. [ Links ]
40. SELLIER C, RAU F, LIU Y, TASSONE F, HUKEMA RK, GATTONI R, ET AL. Sam68 sequestration and partial loss of function are associated with splicing alterations in FXTAS patients. EMBO J. 2010;29:1248-61. [ Links ]
41. DENLI AM, TOPS BBJ, PLASTERK RHA, KETTING RF, HANNON GJ. Processing of primary microRNAs by the Microprocessor complex. Nature 2004;432:231-5. [ Links ]
42. SELLIER C, FREYERMUTH F, TABET R, TRAN T, HE F, RUFFENACH F, ET AL. Sequestration of DROSHA and DGCR8 by expanded CGG RNA repeats alters microRNA processing in fragile X-associated tremor/ataxia syndrome. Cell Rep. 2013, Mar;3(3):869-80 [citado 2014 marzo 24]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3639429&tool=pmcentrez&rendertype=abstract. [ Links ]
43. AMERES SL, ZAMORE PD. Diversifying microRNA sequence and function. Nat Rev Mol Cell Biol. Nature Publishing Group 2013, Aug;14(8):475-88. [citado 2014 marzo 19]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/23800994. [ Links ]
44. WANG D, YANG B. MicroRNA expression detection methods. Vasa; 2010 [citado 2014 abril 17]. Disponible en: http://medcontent.metapress.com/index/A65RM03P4874243N.pdf. [ Links ]
45. RENOUX AJ, TODD PK. Neurodegeneration the RNA way. Progress in Neurobiology; 2012. p. 173-89. [ Links ]
46. KING OD, GITLER AD, SHORTER J. The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease. Brain Research; 2012. p. 61-80. [ Links ]
47. TODD PK, OH S, KRANS A, HE F, SELLIER C, FRAZER M, ET AL. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neurol. 2013;78:440-55. [ Links ]
48. LEEHEY MA, BERRY-KRAVIS E, MIN S-J, HALL DA, RICE CD, ZHANG L, ET AL. Progression of tremor and ataxia in male carriers of the FMR1 premutation. Mov Disord. 2007, Jan;22(2):203-6 [citado 2014 abril 16]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/17133502. [ Links ]
49. HAGERMAN RJ, HALL DA, COFFEY S, LEEHEY M, BOURGEOIS J, ZHANG L, ET AL. Treatment of fragile X-associated tremor ataxia syndrome ( FXTAS ) and related neurological problems 2008;3(2):251-62. [ Links ]
50. HALL DA, BERRY-KRAVIS E, HAGERMAN RJ, HAGERMAN PJ, RICE CD, LEEHEY MA. Symptomatic treatment in the fragile X-associated tremor/ataxia syndrome. Mov Disord. 2006, Oct;21(10):1741-4 [citado 2014 abril 16]. Disponible en: http://www.ncbi.nlm.nih.gov/pubmed/16773616. [ Links ]
51. HAGERMAN RJ, LEAVITT BR, FARZIN F, JACQUEMONT S, GRECO CM, BRUNBERG JA, ET AL. Fragile-X-associated tremor/ataxia syndrome (FXTAS) in females with the FMR1 premutation. Am J Hum Genet. 2004, May;74(5):1051-6. [Internet]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=1181968&tool=pmcentrez&rendertype=abstract. [ Links ]
52. ORTIGAS MC, BOURGEOIS JA, SCHNEIDER A, OLICHNEY J, NGUYEN DV, COGSWELL JB, ET AL. Improving Fragile X-Associated Tremor/Ataxia Syndrome Symptoms With Memantine and Venlafaxine. J Clin Psychopharmacol.; 2010. p. 642-4. [ Links ]
53. SERITAN AL, NGUYEN DV, MU Y, TASSONE F, BOURGEOIS JA, SCHNEIDER A, ET AL. Memantine for fragile X-associated tremor/ataxia syndrome: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry 2014, Mar;75(3):264-71. [ Links ]
54. CAO Z, HULSIZER S, TASSONE F, TANG H, HAGERMAN RJ, ROGAWSKI MA, ET AL. Clustered burst firing in FMR1 premutation hippocampal neurons: amelioration with allopregnanolone. Hum Mol Genet. 2012, Jul;21(13):2923-35 [citado 2014 abril 16]. Disponible en: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3373240&tool=pmcentrez&rendertype=abstract. [ Links ]
