










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.31 no.4 Bogotá Oct. 2015
Trabajo original
Caracterización clínica, bioquímica e imagenológica en una cohorte de pacientes diagnosticados con hiperglicinemia no cetósica clásica: estudio ambispectivo 2000-2014, Medellín, Colombia
Clinical, biochemical and imaging characterization in a cohort of patients diagnosed with classical nonketotic hyperglycinemia: ambispective study 2000-2014, Medellin-Colombia
Juliana Trujillo Gómez (1), Sandra Milena Tobón Carvajal (2), Blair Ortiz Giraldo (3), Sandra Catalina Mesa Restrepo (4), Gabriel Jaime Vélez Rengifo (5), José William Cornejo Ochoa (6)
(1) Pediatra, Universidad de Antioquia, Hospital San Vicente Fundación. Medellín, Colombia.
(2) Pediatra, Universidad de Antioquia, Clínica El Rosario. Medellín, Colombia.
(3) Pediatra y Neurólogo Infantil, Hospital San Vicente Fundación. Medellín, Colombia.
(4) Pediatra y Neuróloga Infantil, Hospital Pablo Tobón Uribe. Medellín, Colombia.
(5) Neurólogo Infantil, Instituto Neurológico de Colombia. Medellín, Colombia.
(6) Neurólogo Infantil; profesor, Universidad de Antioquia. Director Grupo de Investigación Pediaciencias, Msc Epidemiología. Medellín, Colombia.
Recibido: 1/06/15. Aceptado: 3/9/15.
Correspondencia: Juliana Trujillo Gómez: july323@hotmail.com
Resumen
Introducción: La hiperglicinemia no cetósica (HGNC) es un error innato del metabolismo del grupo de las aminoacidopatías, de carácter autosómico recesivo, causado por un defecto en el sistema de clivaje de la glicina. Es una entidad rara y no se conoce su incidencia en Colombia.
Objetivo:Describir características clínicas, bioquímica e imagenológicas en una cohorte de pacientes diagnosticados con hiperglicinemia no cetósica clásica
Materiales y métodos: Estudio de tipo descriptivo, ambispectivo, en el periodo enero 2000-2014, en varios centros de Medellín.
Resultados: Se incluyeron 20 pacientes que cumplían criterios de inclusión, de los 35 pacientes que cumplían con el criterio de búsqueda, en su mayoría de sexo femenino y con un Apgar adecuado al nacer. El promedio de inicio de los síntomas fue de 2,6 días; somnolencia, hipoactividad, apnea, convulsiones y singulto fueron los principales síntomas, y las convulsiones de tipo focal las más frecuentes. La relación glicina LCR/plasma en promedio fue 0,42. El patrón estallido-supresión en el electroencefalograma y la ausencia o retraso en la mielinización de la sustancia blanca supratentorial en la resonancia magnética fueron hallazgos comunes.
Conclusión: La HGNC es frecuente en nuestro medio, por lo cual es necesario que se disponga de pruebas bioquímicas y moleculares necesarias para diagnóstico oportuno, manejo integral y asesoría genética.
Palabras clave: Error innato del metabolismo, estallido-supresión, glicina, hiperglicinemia no cetósica, singulto, somnolencia (DECS).
Summary
Introduction: Nonketotic Hyperglycinemia is an inborn error of metabolism in a group of aminoacidopathies, autosomal recessive, caused by a defect in the system of the glycine cleavage. It is rare, and the incidence is unknown in Colombia.
Objective: To describe clinical, biochemical and imaging characteristics in a cohort of patients diagnosed with classical nonketotic hyperglycinemia.
Materials and methods: This is a descriptive-ambispective study during the period January 2000 - 2014 in some centers of Medellin.
Results: There were 35 patients who met the search criteria and finally 20 patients who met inclusion criteria. We found in this cohort more girls than boys, and most of them with a good APGAR. The average onset of symptoms was 2.6 days, with drowsiness, hypoactivity, apnea, seizures and singultus the main symptoms. The focal seizures were the most frequent type. The average value of CSF glycine to plasma glycine ratio was 0.42. The burst suppression pattern in the EEG and the absence or delayed myelination in the supratentorial white matter on MRI were common findings. All patients received dextromethorphan as part of their treatment and the vast majority of sodium benzoate.
Conclusion: HGNC is common in our environment. It´s necessary to have available biochemical and molecular evidence for timely diagnosis, comprehensive management and genetic counseling.
Key words: Burst suppression pattern, drowsiness, glycin, inborn error of metabolism, non ketotic Hyperglycinemia, singultus (MeSH).
Introducción
La hiperglicinemia no cetósica (HGNC) es un error innato del metabolismo del grupo de las aminoacidopatías, de carácter autosómico recesivo, causado por un defecto en el sistema de clivaje de la glicina (SCG), un aminoácido muy tóxico en el sistema nervioso central (SNC) (1, 2).
Es una entidad rara, con una incidencia reportada de 1 en 250.000 personas en Estados Unidos (1, 3, 4), pero relativamente alta en ciertas áreas, como en Finlandia, donde es de 1 en cada 12.000 (5). En Colombia no se cuenta con datos sobre incidencia de HGNC; sin embargo, se conocen descripciones de casos en Bogotá y el Eje Cafetero (2, 6-8).
En la edad pediátrica existen dos formas de presentación clínica, típica y atípica; en la típica la presentación neonatal es la más frecuente y grave (9). Para el diagnóstico definitivo de esta enfermedad se requiere la cuantificación del sistema de clivaje de glicina en hepatocitos o por método molecular (10, 11), pero son métodos costosos y muestran dificultades técnicas (12, 13), motivos por los que suele realizarse con la relación de glicina LCR/plasma (9) (14), el método más usado en la actualidad. En nuestro medio el diagnóstico es difícil de llevar a cabo, ya que incluso esta última técnica se realiza en muy pocos laboratorios en el país y con altos costos.
El objetivo de este estudio fue describir las características clínicas, imagenológicas y bioquímicas de pacientes diagnosticados con hiperglicinemia no cetósica en Medellín.
Materiales y Métodos
Se efectuó un estudio de tipo descriptivo, ambispectivo, en los hospitales Pablo Tobón Uribe (HPTU), San Vicente Fundación (HUSVF) y el Instituto Neurológico de Colombia. Luego de la aprobación por los comités de Ética del Instituto de Investigaciones de la Facultad de Medicina (Universidad de Antioquia) y de cada una de las instituciones, se revisaron las bases de datos en el periodo enero 2000-2014 para seleccionar los registros que aparecían con los diagnósticos "Trastorno del metabolismo de la glicina (CIE10 E725)" y "Trastorno del metabolismo de los aminoácidos (CIE10 E729)". Entre enero de 2012 y marzo de 2014, se hizo seguimiento a las historias clínicas de los pacientes diagnosticados durante estos años como parte prospectiva del estudio.
Criterios de inclusión y exclusión
Los registros seleccionados se revisaron y se procedió a incluir en el estudio los pacientes que cumplieron los criterios definitivos o probables, los cuales fueron definidos con base en la discusión y consenso por parte de los autores.
Diagnóstico definitivo de HGNC (9, 15, 16)
Pacientes que cumplieran los siguientes tres criterios:
- Inicio de síntomas entre 0-28 días.
- Cuadro clínico compatible con un trastorno del metabolismo de la glicina que incluyera mínimo dos de las siguientes características: letargia progresiva, hipotonía, apneas, singultus, convulsiones de difícil control y retraso en el desarrollo psicomotor.
- Uno o más de los siguientes criterios: relación glicina LCR/plasma > 0,04 o defecto molecular en el gen GLDC, AMT o GCSH.
Diagnóstico probable de HGNC
Pacientes que cumplieran los siguientes tres criterios:
- Inicio de síntomas entre 0-28 días.
- Cuadro clínico compatible con un trastorno del metabolismo de la glicina que incluyera mínimo dos de las siguientes características: letargia progresiva, hipotonía, apneas, singultus, cuadro convulsivo de difícil control y retraso en el desarrollo psicomotor.
- Dos o más de los siguientes criterios: espectroscopia por resonancia magnética sugestiva de HGNC, glicina elevada en LCR > 80 µmol/L, glicina elevada en plasma > 920 µmol/L o patrón EEG de estallido-supresión.
Los valores elegidos de glicina en LCR y plasma se establecieron con base en los datos reportados en algunos estudios de los pacientes con diagnóstico definitivo de HGNC neonatal (9, 15, 16), con el fin de aumentar la especificidad de los casos que no contaron con el método diagnóstico de elección.
Se excluyeron los pacientes que tenían alguna acidemia orgánica o cuya información estaba incompleta en los registros.
Se revisó extensamente cada una de las historias clínicas y se extrajo información relacionada con aspectos clínicos y ayudas diagnósticas de cada paciente en un formulario diseñado previamente para este fin.
Los datos se ingresaron en una base de datos de Excel 2010. Las variables de tipo cualitativo se analizaron como frecuencias y proporciones. Las variables cuantitativas se analizaron como promedios y en algunos casos como medianas y modas. Los datos obtenidos se organizaron como tablas agrupadas según las características clínicas, imagenológicas y de laboratorio.
Resultados
Se encontraron 35 pacientes con diagnóstico de HGNC; de estos se excluyeron 15 registros por información insuficiente en la historia clínica; finalmente, se incluyeron 20 casos en el estudio.
De las historias incluidas, 14 (70%) fueron mujeres y 6 (30%) hombres, procedentes en su mayoría del valle de Aburrá (55%) y el oriente de Antioquia (35%).
Solo 4 pacientes (20%) tenían antecedente de consanguinidad en los padres y 4 (20%) antecedente familiar de muerte en la etapa neonatal.
El promedio de edad gestacional al nacimiento fue de 38 semanas, con un peso promedio de 3.110 gramos y una talla de 49,5 cm. El 90% de los pacientes tuvo un puntaje Apgar adecuado al minuto y a los cinco minutos; solo 2 de los pacientes (10%) requirieron ventilación mecánica al nacer.
El promedio de inicio de los síntomas fue de 2,6 días. El síntoma más frecuente al inicio del cuadro clínico fue somnolencia (30%), seguido por hipoactividad (20%) (Tabla 1). Durante la evolución todos los pacientes presentaron alteraciones en el reflejo de succión, en el llanto y en el estado de conciencia, además de hipotonía, hipoactividad y convulsiones. El singultus estuvo presente en el 50% de los pacientes (Tabla 2).
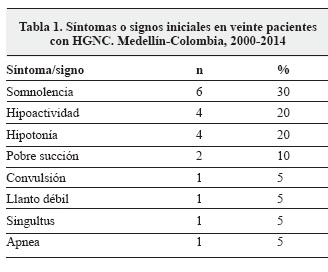

Con relación a las convulsiones durante la evolución de la enfermedad, las focales fueron las más frecuentes (80%), seguidas de las crisis tonicoclónicas generalizadas (60%), mioclónicas (60%) y espasmos infantiles (60%).
La cuantificación de glicina en plasma fue informada en 12 de los pacientes, con un valor promedio de 1.488 (DE 1.019,64), mientras que la glicina en LCR solo estuvo disponible en 10 casos, con un valor promedio de 563 (DE 1.129,35). El promedio de la relación glicina LCR/plasma fue de 0,42 (DE 0,49).
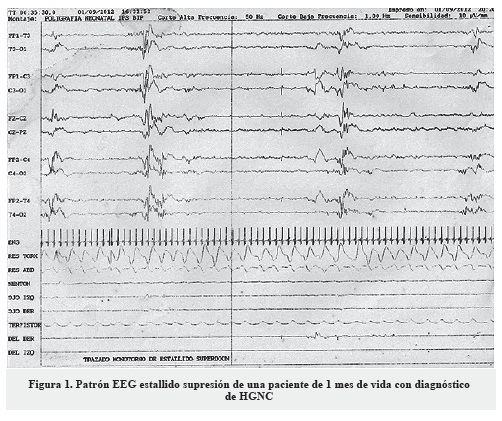
El patrón estallido-supresión en el electroencefalograma se encontró en el 78%, seguido de la hipsarritmia en el 36%. En 17 de los pacientes estuvo disponible información acerca de la resonancia magnética cerebral, la cual se encuentra detallada en la Tabla 3.

Todos los pacientes recibieron dextrometorfano como parte de su tratamiento y en su gran mayoría benzoato de sodio (95%). El 90% de los pacientes requirió más de un anticonvulsivante para el control de sus crisis. En general se logró disminuir el número de episodios convulsivos en un 82% de los pacientes y mejorar el estado de conciencia en el 70% de ellos. Siete de los pacientes (35%) requirieron gastrostomía. El 60% de ellos recibió de forma más constante fórmula láctea especial libre de aminoácidos durante su evolución.
Todos los pacientes que asistieron a controles a la institución después de su egreso presentaban compromiso neurológico severo a nivel cognitivo y motor.
Discusión
La HGNC es un error innato del metabolismo que se manifiesta con frecuencia en la etapa neonatal, y los que sobreviven quedan con un importante compromiso del neurodesarrollo y epilepsia refractaria (9, 14).
En la revisión de la literatura no se encontraron estudios descriptivos en Colombia que reportaran un número similar de pacientes con HGNC, tal como lo presentado en este estudio.
En cuanto a la procedencia de los pacientes, se observa cómo el 35% viene del oriente antioqueño, lo cual puede explicarse por la alta tasa de endogamia descrita en las familias de esta región y que incrementa el riesgo de presentación de enfermedades genéticas con patrón de transmisión autosómico recesivo (17). De los pacientes analizados, en el 25% hubo algún tipo de consangunidad en las familias, pero es probable que esta cifra sea mayor si se realizara de manera rigurosa el análisis genealógico.
Por el tamaño de la muestra y el tipo de estudio efectuado, no fue posible demostrar un curso más grave de la enfermedad en las niñas como lo describen algunos estudios (18).
De acuerdo con lo informado en la literatura médica, en general son embarazos y partos normales (18). El 90% de los pacientes de nuestra investigación fueron nacimientos a término, con antropometría adecuada y adaptación neonatal espontánea.
Los síntomas se presentaron en la mayoría de pacientes en las primeras 24-72 horas de vida, encontrándose como los hallazgos clínicos iniciales más relevantes: somnolencia, pobre succión, apneas y llanto débil/ausente; el 65% evolucionó hacia la depresión respiratoria y/o apneas persistentes que los llevaron a requerir ventilación mecánica en los primeros días de vida. Estos hallazgos están acorde a lo reportado en las dos series grandes de Carson NAJ y Hoover-Fong et al. (14, 18). El singultus se identificó en el 50% de los pacientes, síntoma reportado con frecuencia en esta entidad y explicado por el efecto inhibitorio de la glicina en el tallo cerebral y la médula espinal (15).
Las convulsiones se presentaron en el 100% de los pacientes de nuestro estudio. Según la literatura, el tipo de convulsión más observado es el mioclónico, contrastando con lo encontrado en esta investigación, en la que predominaron las de tipo focal (80%), seguidas de las tonicoclónicas generalizadas, mioclónicas y de tipo espasmos infantiles, reportadas en el 60%. Se ha descrito que en la HGNC neonatal las convulsiones pueden aparecer en los primeros 3 meses de vida (19), pero en otros estudios incluso mencionan que pueden aparecer mucho más tarde (18). El 90% tuvo convulsiones de difícil control, lo cual ha sido clásicamente descrito por el efecto de la glicina sobre los receptores NMDA, que son de tipo excitatorio (20).
En el presente trabajo 8 de 20 pacientes contaron con el resultado de la relación glicina LCR/plasma, con un promedio en esta relación de 0,42, una cifra mucho mayor que la reportada por Hoover-Fong et al. (2003); en los demás pacientes el diagnóstico se realizó sumando otros criterios (glicina elevada en LCR o plasma, patrón EEG estallido-supresión y/o espectroscopia con pico de glicina); en ninguno se hizo estudio enzimático, hepático, ni molecular.
El patrón electroencefalográfico de estallido-supresión, si bien no es específico de la HGNC, sí es una característica importante para el diagnóstico, ya que se encuentra con frecuencia en los primeros meses de vida de los pacientes con HGNC (19) (Figura 1). En esta serie se encontró en el 78% de los pacientes y el patrón de hipsarritmia en el 36%.
Con relación a la evolución temporal de los pacientes que sobrevivieron, todos presentaron retraso grave del neurodesarrollo, con compromiso motor y cognitivo asimismo grave, similar a como lo han reseñado otros estudios (9, 14, 19).
Uno de los pacientes evaluados presentó de forma concomitante defecto de la betaoxidación; no se ha encontrado en la literatura asociación entre estas dos entidades.
En cuanto al tratamiento, todos los pacientes recibieron dextrometorfano y el 95% benzoato de sodio, los dos medicamentos más recomendados actualmente para la terapia de esta enfermedad (18, 21, 22); en general hay mejoría del estado de conciencia y de las convulsiones en más de la mitad de los pacientes, aunque sin impacto en el neurodesarrollo, tal como se ha descrito (19, 23).
Se encontró marcada intolerancia gástrica con el benzoato de sodio que condujo a la suspensión de este en varios de ellos. Se describe que la gastritis es un efecto secundario encontrado con frecuencia, por ello la importancia de agregar un protector de la mucosa gástrica (21).
El 65% de nuestros pacientes recibieron fórmula láctea especial -libre de glicina- durante la evolución de la enfermedad. Si bien no contamos con los suficientes datos para determinar el efecto de esta medida en nuestros pacientes, varios artículos sustentan la importancia de una restricción dietaria adecuada junto con el manejo farmacológico, de efectos benéficos en cuanto a la mejoría de las convulsiones y el estado de conciencia (19, 22).
Cabe recalcar que en el estudio se evidenciaron dificultades que reflejan el estado actual de nuestra región para el diagnóstico oportuno de los trastornos metabólicos. En el caso de la HGNC, no contamos en la ciudad con un laboratorio que realice la cuantificación de glicina en LCR y plasma, a lo que se suman los obstáculos que ofrecen las aseguradoras para completar los estudios de las consideradas "enfermedades raras", entre las cuales se encuentran los errores innatos del metabolismo; por esta razón fue necesario incluir en este trabajo un grupo de pacientes sin el diagnóstico bioquímico de elección (diagnóstico probable de HGNC). La información insuficiente en algunas historias clínicas fue otro factor limitante del estudio, y el principal para excluir varios pacientes del análisis realizado.
Finalmente, es posible que el número de pacientes en el periodo de estudio sea mayor y fuesen evaluados en alguna de las instituciones que no se incluyeron en él; por otro lado, y de mayor relevancia, es la alta probabilidad de que a menudo el diagnóstico de HGNC sea omitido, especialmente en los casos neonatales severos que fallecen bajo otro tipo de diagnóstico y en los casos atípicos que se presentan luego de la etapa neonatal.
Conclusiones
La HGNC es una enfermedad neurometabólica frecuente en nuestro medio.
La presencia de síntomas como somnolencia, hipoactividad, pobre succión y apneas en las primeras horas de vida debe incrementar la sospecha de esta enfermedad; hallazgos como el patrón de estallido-supresión en el EEG, el retraso de mielinización de la sustancia blanca supratentorial y el pico de glicina en la espectroscopia apoyan el diagnóstico, el cual debe corroborarse con la relación de glicina LCR/plasma, el estudio molecular, o el estudio enzimático.
Es necesario que en el país se disponga de las pruebas bioquímicas y moleculares necesarias para el diagnóstico oportuno de HGNC, con el fin de ofrecer una intervención integral y realizar la asesoría genética respectiva.
Agradecimientos. Al Hospital Pablo Tobón Uribe, al Hospital Universitario San Vicente Fundación y al Instituto Neurológico de Colombia, por permitirnos revisar sus bases de datos y tener acceso a sus historias clínicas.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Referencias
1. BERMÚDEZ M, ARTEAGA C, CIFUENTES Y. Hiperglicinemia no cetósica (HGNC). Forma típica y atípica. Presentación de casos diagnosticados en Colombia. Rev. Fac. Med. 2007;55(2):126-34. [ Links ]
2. CIFUENTES Y, BERMÚDEZ M, ARTEAGA C. Encefalopatía neonatal. Algo más que asfixia al nacer. Presentación de casos. Rev. Fac. Med. 2007;55(2):126-34. [ Links ]
3. BERRY GT. Inborn errors of carbohydrate, ammonia, amino acid, and organic acid metabolism. In: Teaeusch, Ballard, Gleason, eds. Avery's diseases of the newborn. Elsevier; 2005. p. 227-57. [ Links ]
4. BHAMKAR RP, COLACO P. Neonatal nonketotic hyperglycinemia. Indian journal of pediatrics 2007 Dec;74(12):1124-6. [ Links ]
5. VON WENDT L, HIRVASNIEMI A, SIMILÄ S. Nonketotic hyperglycinemia. A genetic study of 13 Finnish families. Clinical genetics 1979 May;15(5):411-7. [ Links ]
6. PERNA JA, GUTIÉRREZ C. PDH. Hiperglicinemia no cetósica. Repertorio de Medicina y Cirugía 2001;10(3):42-6. [ Links ]
7. BERMÚDEZ M, ARTEAGA C, CIFUENTES Y, ESPINOSA E, URIBE A, BARRERA L, ET AL. Hiperglicinemia no cetósica (HGNC). Forma típica y atípica: casos clínicos diagnosticados en Colombia. Pediatría 2001;36(2):123-9. [ Links ]
8. URIBE A, ESPAÑA M, MURILLO P. Doce años de investigación (1996-2008): tamizaje de alto riesgo de errores innatos del metabolismo de aparición temprana. Memorias - I Congreso Latinoamericano de Genética Humana / IX Congreso Colombiano de Genética; 2008. p. M6. [ Links ]
9. VAN HOVE J, COUGHLIN C, MBE AND GS. Glycine Encephalopathy. GeneReviews®. 2013. Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1357/ [ Links ]
10. HAMOSH A, SCHARER G, VAN HOVE J. Glycine Encephalopathy. GeneReviews. 2009 Disponible en: http://www.ncbi.nlm.nih.gov/books/NBK1116/ (Último acceso: marzo 5 de 2012). [ Links ]
11. APPLEGARTH DA, TOONE JR. Nonketotic hyperglycinemia (glycine encephalopathy): laboratory diagnosis. Molecular genetics and metabolism 2001;74(1-2):139-46. [ Links ]
12. TOONE JR, APPLEGARTH DA, COULTER-MACKIE MB, JAMES ER. Recurrent mutations in P- and T-proteins of the glycine cleavage complex and a novel T-protein mutation (N145I): a strategy for the molecular investigation of patients with nonketotic hyperglycinemia (NKH). Molecular genetics and metabolism 2001 Apr;72(4):322-5. [ Links ]
13. TOONE JR, APPLEGARTH DA, COULTER-MACKIE MB, JAMES ER. Biochemical and molecular investigations of patients with nonketotic hyperglycinemia. Molecular genetics and metabolism 2000 Jun;70(2):116-21. [ Links ]
14. HOOVER-FONG JE, SHAH S, VAN HOVE JLK, APPLEGARTH D, TOONE J, HAMOSH A. Natural history of nonketotic hyperglycinemia in 65 patients. Neurology 2004 Nov 23;63(10):1847-53. [ Links ]
15. HAMOSH A, JOHNSTON MV. Part 8: Amino acids Chapter 90. Nonketotic Hyperglycinemia In: Hamosh A, Johnston MV. Prenatal Diagnosis. p. 1-30. [ Links ]
16. HAYASAKA K, TADA K, FUEKI N, NAKAMURA Y, NYHAN WL, SCHMIDT K, ET AL. Nonketotic hyperglycinemia: analyses of glycine cleavage system in typical and atypical cases. The Journal of pediatrics 1987 Jun;110(6):873-7. [ Links ]
17. BEDOYA G, GARCÍA J, MONTOYA P, ROJAS W, EUGENIA M, SOTO I, ET AL. Análisis de isonimia entre poblaciones del noroeste de Colombia. Biomédica 2006;26:538-45. [ Links ]
18. CARSON NAJ for NW. 17th Symposium of SSIEM. Non-ketotic Hyperglycinaemia - A Review of 70 Patients. J. Inher. Metab. Dis. 1982;2:126-8. [ Links ]
19. HENNERMANN JB. Clinical variability in glycine encephalopathy. Future Neurol. 2006;1:621-30. [ Links ]
20. SUZUKI Y, KURE S, OOTA M, HINO H, FUKUDA M. Nonketotic hyperglycinemia: proposal of a diagnostic and treatment strategy. Pediatric neurology 2010 Sep;43(3):221-4. [ Links ]
21. HAMOSH A, MAHER JF, BELLUS G A, RASMUSSEN S A, JOHNSTON MV. Long-term use of high-dose benzoate and dextromethorphan for the treatment of nonketotic hyperglycinemia. The Journal of pediatrics 1998 Apr;132(4):709-13. [ Links ]
22. VAN HOVE JLK, VANDE KERCKHOVE K HJ, MAHIEU V, DECLERCQ P, MERTENS S. Benzoate treatment and the glycine index in nonketotic hyperglycinaemia. J Inherit Metab Dis. 2005;28(5):651-63. [ Links ]
23. LU FL, WANG PJ, HWU WL, TSOU YAU KI, WANG TR. Neonatal type of nonketotic hyperglycinemia. Pediatric neurology 1999 Apr;20(4):295-300. [ Links ]
