










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.32 no.1 Bogotá Jan./Mar. 2016
Caso clínico
Distrofia muscular oculofaríngea. Presentación de un caso particular e importancia de los hallazgos electromiográficos
Oculopharyngeal muscular dystrophy. Presentation of a peculiar case and importance of electromyographic findings
Antonio Díaz Negrillo (1)
(1) Unidad de Neurofisiología Clínica, Hospital Universitario Infanta Elena. Valdemoro, Madrid, España.
Recibido: 15/12/15. Aceptado: 5/02/16.
Correspondencia: Antonio Díaz Negrillo.: antoniodnegrillo@yahoo.es.
Resumen
La distrofia muscular oculofaríngea es un raro trastorno genético caracterizado clínicamente por ptosis palpebral y dificultades deglutorias. Con este trabajo se pretende hacer una descripción detallada de un caso particular, poniendo de manifiesto la relevancia de los hallazgos electromiográficos, además se propone realizar una exhaustiva revisión bibliográfica sobre el tema.
Fue un estudio clínico, neurofisiológico y molecular de una paciente de 82 años remitida a la Unidad de Neurofisiología Clínica por ptosis palpebral bilateral y disfagia progresiva.
El estudio electromiográfico objetivó la existencia de una miopatía de predominio facial y el estudio molecular confirmó la existencia de una expansión patológica de 12 repeticiones en el tracto poli-alanina del gen PABPN1.
La distrofia muscular oculofaríngea es una enfermedad infradiagnosticada que puede pasar desapercibida, simulando otros procesos patológicos. Un riguroso estudio neurofisiológico es fundamental para llevar a cabo un diagnóstico de sospecha así como para orientar el posterior diagnóstico molecular.
Palabras clave: Distrofia Muscular Oculofaríngea. Miopatía. Electromiografía.(DeCS).
Summary
Oculopharyngeal Muscular Dystrophy is a rare genetic disorder characterized clinically by ptosis and deglutition difficulties. This work aims to give a detailed description of a particular case highlighting the relevance of the electromyographic findings in addition to a comprehensive literature review on the topic.
Clinical, neurophysiological and molecular study of 82 years old patient referred to Clinical Neurophysiology Unit for progressive bilateral ptosis and dysphagia.
The electromyographic study aimed the existence of a facial myopathy prevalence and molecular study confirmed the existence of a pathological expansion of 12 repetitions in the poly-alanine tract PABPN1 gene. Conclusions: Oculopharyngeal Muscular Dystrophy is an underdiagnosed disease that may go unnoticed by simulating other pathological processes. A rigorous neurophysiological study is essential to conduct a suspected diagnosis and to guide further molecular diagnostics.
Key words: Oculopharyngeal Muscular Dystrophy. Myopathy. Electromyography (MeSH).
Introducción
La distrofia muscular oculofaríngea es una alteración genética poco frecuente, que se manifiesta clínicamente por ptosis palpebral, disfagia y distrofia muscular. Otros síntomas menos frecuentes son debilidad lingual, trastornos fonatorios, debilidad de la musculatura facial o debilidad de las extremidades (1). Esta enfermedad fue descrita inicialmente por Hutchinson en 1879 (2) y posteriormente por Taylor en 1915 (3). Se trata de una patología de carácter progresivo e inicio en la edad adulta, cuya base etiopatogénica reside en una mutación en la expansión de un tracto de poli-alanina del gen PABPN1 (4). El patrón de herencia más frecuente es el autosómico dominante, aunque también se han observado formas recesivas, con un curso más leve e inicio más tardío. En los casos de herencia autosómica dominante cada descendiente de una persona afectada tiene un 50% de probabilidades de heredar el alelo mutado. Sin embargo, en los casos de herencia autosómica recesiva cada descendiente de un individuo afectado tiene un 25% de probabilidad de estar afectado, un 50% de probabilidad de ser portador asintomático así como un 25% de posibilidades de estar afectado y no ser portador (5). El diagnóstico de sospecha se lleva a cabo atendiendo los criterios clínicos y el diagnóstico de confirmación se sustenta en pruebas genéticas moleculares. En la actualidad no se dispone de tratamiento efectivo. Las medidas aplicadas son fundamentalmente de soporte, nutricionales y quirúrgicas (6).
Presentamos el caso de una señora de 82 años enviada a la Unidad de Neurofisiología Clínica por sospecha de miastenia gravis y cuyo estudio electromiográfico fue fundamental para orientar el diagnóstico hacia una distrofia oculofaríngea.
Presentación del caso
Paciente mujer de 82 años, con antecedentes de osteoporosis e histerectomía, remitida a la Unidad de Neurofisiología Clínica por cuadro de ptosis bilateral lentamente progresiva y dificultad ocasional para la deglución. En la exploración física la paciente presenta un claro enoftalmos acompañado de ptosis palpebral bilateral, ligeramente asimétrica (mayor en el párpado izquierdo), con imposibilidad para la supraversión de la mirada. La ptosis palpebral empeoró con la contracción repetida del músculo orbicular de los ojos. El test de Cogan fue poco valorable. Además presentó una discreta hipofonía que empeoró levemente al contar hasta 50. La paciente se quejó de dificultad en la masticación y debilidad en las cuatro extremidades, de predominio en musculatura proximal. Se le realizó una batería analítica compuesta por hemograma y bioquímica sanguíneas y de orina, estudio de coagulación, estudio enzimático con enzimas hepáticas, lactato deshidrogenasa, aldolasa y creatín quinasa, proteinograma, perfil tiroideo, Vitamina B12 y ácido fólico, hemoglobina glicosilada, serologías (VIH, borrelia), pruebas treponémicas (ELISA) así como estudio de anticuerpos antinucleares, anticuerpos anticitoplasma de neutrófilo y receptores de acetilcolina.
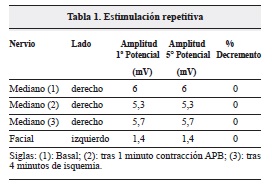
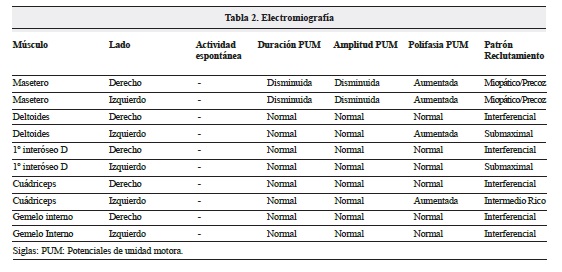
Todos los resultados fueron normales. Se llevó a cabo un estudio vídeo-endoscópico, se evidenció un trastorno de la deglución. Se realizó un completo estudio de conducción nerviosa en el que se valoró la conducción de ambos nervios medianos, cubitales, peroneales profundos, tibiales (con ondas F) y surales así como una estimulación repetitiva del nervio mediano derecho en condiciones basales, tras 1 minuto de contracción mantenida del músculo separador corto del pulgar derecho y tras 4 minutos de isquemia de la extremidad superior derecha. Se amplió el estudio realizando una estimulación repetitiva del nervio facial izquierdo ("Tabla 1). Por otro lado, se valoró la actividad electromiográfica de los músculos maseteros, deltoides, primeros interóseos dorsales, cuádriceps y gemelos internos de ambos lados. La paciente presentó una estatura de 1,65 metros y las mediciones se realizaron a una temperatura ambiental de 22ºC. El estudio neurofisiológico evidenció unos hallazgos en la conducción nerviosa y en la estimulación repetitiva normales, siendo patológicos los hallazgos electromiográficos (Tabla 2, Figura 1).
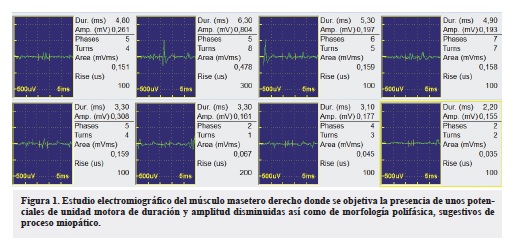
Estos hallazgos neurofisiológicos sugirieron la existencia de una miopatía de naturaleza distrófica, no miotónica, y de localización casi exclusivamente facial. Ante la sospecha diagnóstica de una posible distrofia muscular oculofaríngea, se realizó un estudio molecular, siguiendo el procedimiento que se describe a continuación:
Estudio molecular
La metodología para llevar a cabo el estudio molecular fue la siguiente:
1. Extracción del ADN (con Bio robot EZ1 Qiagen)
2. Amplificación por Reacción en cadena de la polimerasa (PCR) de la región repetida GCN que codifica para un tracto de poli-alanina.
3. Electroforesis de los productos de PCR (con Abiprism 3130: GeneMapper V 4.0)
Los resultados del estudio molecular fueron los siguientes:
Paciente portadora de un alelo normal de 10 repeticiones y otro que presenta expansión patológica de 12 repeticiones en el tracto poli-alanina (GCN) del gen PABP1 responsable de la distrofia muscular orofaríngea.
Discusión
La distrofia muscular culofaríngea (DOF) es una enfermedad de origen genético que afecta fundamentalmente al músculo estriado. Aunque las primeras descripciones fueron llevadas a cabo por Hutchinson en 1879 y posteriormente por Taylor en 1915, no fue hasta 1998 cuando Brais describió la alteración genética causante de esta enfermedad (4). En términos epidemiológicos, se ha visto que la DOF se presenta más frecuentemente en población franco-canadiense. De hecho, el mayor núcleo de prevalencia se encuentra en Quebec. Se conoce la existencia de una familia de emigrantes franceses (Zachaire Cloutier y Saint Dupont), que en el año 1648 se instalaron en Quebec, propagando la enfermedad en la descendencia (7). También es frecuente en la población judía procedente de judíos Bukhara que emigraron a Israel procedentes de Uzbekistán (8). En el caso de nuestra paciente, se desconoce tenía antecedentes franceses o judíos.
La alteración genética se encuentra localizada en el cromosoma 14. La mutación responsable es una expansión anómala de un tracto de poli-alanina (GCN) ubicada en el primer exón del gen PABNP1. Este gen codifica una proteína exclusivamente intra-celular con alta expresión en el músculo. Se cree que la proteína mutada no puede ser degradada y por tanto se produce su acumulación intracelular, con la consecuente alteración del metabolismo celular del miocito hasta provocar su destrucción (9). La población normal presenta un número de hasta 10 repeticiones. La enfermedad se suele heredar, como en nuestro caso, con carácter autosómico dominante, aunque hay formas recesivas descritas. La secuencia poli-alanina (GCN) puede sufrir pequeñas expansiones patológicas de entre las 12 y 17 repeticiones para las formas dominantes y 11 para las formas recesivas, con un curso más leve e inicio más tardío. En términos porcentuales, el número de repeticiones patológicas más frecuente es de 13 (40%) (10). El caso que nos ocupa, al presentar una expansión patológica de 12 repeticiones, tiene la particularidad de ser de los menos frecuentes (5%). La descendencia presenta un riesgo de un 50% de heredar la mutación. Sospechamos que la hija de la paciente ha heredado la mutación, dado que padece una sintomatología similar a la descrita en la madre (si bien una disfagia menos acentuada) y ya ha sido operada de blefaroplastia (actualmente se encuentra a la espera del resultado del estudio genético molecular). Desde el punto de vista clínico, los dos síntomas típicos y más relevantes de la enfermedad son la ptosis palpebral y disfagia. La edad media de aparición de la sintomatología principal suele ser entre los 45-50 años. También puede aparecer como sintomatología acompañante, aunque menos frecuente, debilidad de la musculatura facial, trastornos de la fonación o debilidad muscular a otros niveles. De forma excepcional puede verse afectado el Sistema Nervioso (11). Nuestro caso presentó los síntomas típicos, y los anteriormente descritos. Los tres criterios clínicos para el diagnóstico de sospecha de la distrofia muscular oculofaríngea son los siguientes: 1. Historia familiar positiva en al menos dos o más generaciones. 2. Presencia de ptosis palpebral, definida como separación vertical de al menos una hendidura palpebral que mide menos de 8 mm en reposo.
También se incluye el antecedente de cirugía correctiva-blefaroplastia. 3. Presencia de disfagia entendida como un tiempo de deglución mayor a 7 segundos, cuando se bebe 80 ml de agua enfriada con hielo. Estos tres criterios clínicos son válidos tanto para la forma autosómica dominante como para la recesiva, si bien los signos y síntomas de esta última son más leves y más tardíos en su aparición (12). En este último caso, la historia familiar debe ser congruente con una herencia autosómica recesiva. Los hallazgos analíticos suelen ser inespecíficos. Se han descrito casos de DOF en los que se hallaron concentraciones de creatin quinasa sérica elevada hasta siete veces por encima del límite alto de la normalidad. Sin embargo, lo habitual es encontrar unos niveles séricos de dicha enzima normales o discretamente elevados. Por lo tanto, actualmente dicho hallazgo no se considera relevante ni para el diagnóstico ni para el seguimiento de la enfermedad (13). Antes de la introducción de las pruebas genéticas moleculares, la biopsia muscular era una herramienta muy importante en el diagnóstico de la DOF. Los hallazgos anatomopatológicos más representativos de la DOF son la presencia de inclusiones intranucleares específicas en los miocitos así como los cambios distróficos, que se manifiestan con las transformaciones en el diámetro de las fibras musculares, atrofia de fibras, fibras rojas rasgadas y presencia de vacuolas intracelulares (14). En nuestra paciente no se practicó biopsia. De hecho, en la actualidad la biopsia muscular solo es utilizada en aquellos pacientes que por estudio genético manifiestan dos alelos normales PABPN1. El estudio electromiográfico de los músculos afectos generalmente revela datos de un proceso miopático (15). Habitualmente no se le ha dado mucha importancia a los hallazgos neurofisiológicos. El caso que presentamos pone de manifiesto que una exploración electroneurográfica y electromiográfica rigurosa nos puede aportar información relevante y decisiva para el diagnóstico tal como la naturaleza miopática del proceso, el carácter distrófico y la localización anatómica del mismo. En esta línea los estudios neurofisiológicos son de gran ayuda para la orientación hacia el posterior estudio molecular. El diagnóstico de confirmación se lleva a cabo mediante pruebas genéticas moleculares en las que se extrae el ADN, se amplifica (generalmente mediante una reacción en cadena de la polimerasa) y posteriormente se lleva a cabo una electroforesis del producto amplificado (16-17). Ante la sospecha de una posible DOF se deben incluir dentro del diagnóstico diferencial todas aquellas enfermedades neuromusculares de aparición tardía y que cursen con ptosis y/o disfagia. Además, debemos prestar especial consideración a las distrofias miotónicas, miastenia gravis y otras enfermedades de la unión neuromuscular, miopatías inflamatorias, tiroideas y mitocondriales o neuropatías hereditarias de predominio motor (18). En esta línea también son de suma utilidad el estudio electroneurográfico y electromiográfico, esto nos permiten realizar una discriminación y orientación razonable de la naturaleza del proceso. Por ello, como muestra el caso presentado, aconsejamos que en aquellos pacientes en los que exista una sintomatología sugestiva, se lleve a cabo un estudio neurofisiológico reglado y riguroso. Dicha exploración debe incluir un estudio electroneurográfico con realización de estimulación repetitiva o estudio de fibra aislada, valoración de la velocidad de conducción nerviosa en nervios motores y sensitivos de las cuatro extremidades, estudiar respuestas tardías (ondas F) de al menos dos nervios y completar la exploración con un estudio electromiográfico de musculatura proximal y distal de extremidades, así como de musculatura craneal y/o cervical. Ello posibilitará, como se ha mostrado en este trabajo, conseguir una orientación diagnóstica más rigurosa, mejorando sustancialmente la sensibilidad y especificidad del proceso del diagnóstico general.
Conflicto de intereses
Los autores declaran no tener conflicto de intereses.
Referencias
1. BRAIS B, ROULEAU GA, BOUCHARD JP, FARDEAU M, TOME FM. Oculopharyngeal muscular dystrophy. Semin Neurol. 1999; 19 (1): 59-66. [ Links ]
2. HUTCHINSON J. On Ophthalmoplegia Externa, or Symmetrical Immobility (partial) of the Eyes, with Ptosis. Med Chir Trans. 1879; 62: 307-29. [ Links ]
3. TAYLOR EW. Progressive vagus-glossopharyngeal paralysis with ptosis: contribution to group of family disease. J Nerv Ment Disord 1915; 42: 129-39. [ Links ]
4. BRAIS B, BOUCHARD JP, ROCHEFORT DL ET AL. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nature Genet 1998; 18 (2): 164-67. [ Links ]
5. HARISH P, MALERBA A, DICKSON G, BACHTARZI H. Progress on gene therapy, cell therapy, and pharmacological strategies toward the treatment of oculopharyngeal muscular dystrophy. Hum Gene Ther. 2015; 26(5): 286-92. [ Links ]
6. HERNÁNDEZ-MONTERO E, MESA-MARRERO M, FRÍAS-BERZOSA BD. ET AL. Oculopharyngeal muscular dystrophy: a case report and review of the literature. Acta Otorrinolaringol Esp. 2012 63(6):482-4. [ Links ]
7. BARBEAU A. The syndrome of hereditary late-onset ptosis and dysphagia in French Canada. In: Kuhn E, ed. Symposium über Progressive Muskeldystrophie. Berlin, Germany: Springer-Verlag. 1996. P. 102-109. [ Links ]
8. BLUMEN SC, SADEH M, KORCZYN AD, ROUCHE A, NISIPEANU P, ASHEROV A, TOME FM. Intranuclear inclusions in oculopharyngeal muscular dystrophy among Bukhara Jews. Neurology. 1996; 46 (5): 1324-8. [ Links ]
9. CHARTIER A, KLEIN P, PIERSON S, ET AL. Mitochondrial dysfunction reveals the role of mRNA poly(A) tail regulation in oculopharyngeal muscular dystrophy pathogenesis. PLoS Genet. 2015 Mar 27;11 (3). [ Links ]
10. JOUAN L, ROCHEFORD D, SZUTO A, ET AL. An 18 alanine repeat in a severe form of oculopharyngeal muscular dystrophy. Can J Neurol Sci. 2014; 41(4): 508-11. [ Links ]
11. LUIGETTI M, LO MONACO M, MIRABELLA M, ET AL. Oculopharyngeal muscular dystrophy: Clinical and neurophysiological features. Clin Neurophysiol. 2015; 20 (12): 2406-8. [ Links ]
12. RAZ V, BUTLER-BROWNE G, VAN ENGELEN B, ET AL. 191st ENMC international workshop: recent advances in oculopharyngeal muscular dystrophy research: from bench to bedside 8-10 June 2012, Naarden, The Netherlands. Neuromuscul Disord. 2013;23 (6): 516-23. [ Links ]
13. BRAIS B. Oculopharyngeal muscular dystrophy. Handb Clin Neurol. 2011; 101: 181-92. [ Links ]
14. GIDARO T, NEGRONI E, PÉRIÉ S, ET AL. Atrophy, fibrosis, and increased PAX7-positive cells in pharyngeal muscles of oculopharyngeal muscular dystrophy patients. J Neuropathol Exp Neurol. 2013; 72 (3): 234-43. [ Links ]
15. OSKARSSON B, RINGEL SP. Oculopharyngeal muscular dystrophy as a cause of progression of weakness in antibody positive myasthenia gravis. Neuromuscul Disord. 2013; 23 (4): 316-8. [ Links ]
16. TONDO M, GÁMEZ J, GUTIÉRREZ-RIVAS E, MEDEL-JIMÉNEZ R, ET AL. Genotype and phenotype study of 34 Spanish patients diagnosed with oculopharyngeal muscular dystrophy. J Neurol. 2012; 259 (8): 1546-52. [ Links ]
17. YOU P, MA Q, TAO T. Gene diagnosis of oculopharyngeal muscular dystrophy in a Chinese family by a GeneScan method. J Clin Lab Anal. 2010; 24 (6): 422- 5. [ Links ]
18. JONES LK JR, HARPER CM. Clinical and electrophysiologic features of oculopharyngeal muscular dystrophy: lack of evidence for an associated peripheral neuropathy. Clin Neurophysiol. 2010; 121 (6): 870-3. [ Links ]
