










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.32 no.2 Bogotá Apr./June 2016
Caso clínico
Revisión de Creutzfeld-Jakob a propósito de dos casos clínicos en el Hospital Universitario San Ignacio
Creutzfeldt-Jakob review based on two clinical cases in Hospital Universitario San Ignacio
Felipe Pretelt (•2016) (1), Neiby Rivera (2), Valentina Ursida (3), Catherin Tovar (3), Ayelet Ramírez (3)
(1) Médico neurólogo, Unidad de Neurología, Pontificia Universidad Javeriana, Bogotá, Colombia
(2) Residente de Neurología, Pontificia Universidad Javeriana, Bogotá, Colombia
(3) Estudiante de Medicina, Pontificia Universidad Javeriana, Bogotá, Colombia.
Recibido: 11/11/15. Aceptado: 22/4/16.
Correspondencia: Ayelet Ramírez: ayeletramirezco@gmail.com
Resumen
La enfermedad de Creutzfeld-Jakob es una patología neurodegenerativa fatal e intratable, que hace parte de las denominadas encefalopatías espongiformes y se produce por la acumulación anormal de la PrP (proteína priónica patogénica),denominada PrPsc, a nivel del sistema nervioso central. La enfermedad priónica humana más común es la forma esporádica de la enfermedad de Creutzfeld-Jakob, cuya aparición se ha relacionado con los efectos ambientales desconocidos o los sucesos aleatorios y genéticos, que resultan en la producción espontánea de PrP en el cerebro.
A continuación se presentan dos casos clínicos de dos mujeres que consultan al servicio de urgencias del Hospital Universitario San Ignacio, en quienes se sospechó encefalopatía rápidamente progresiva, compatible con enfermedad de Creutzfeld-Jakob.
Palabras clave: enfermedad de Creutzfeld-Jakob, proteína 14-3-3, proteína Tau, prionopatía esporádica, demencia rápidamente progresiva, DWI (diffusion weighted imaging) (DeCS).
Summary
Creutzfeldt-Jakob disease is a fatal and untreatable neurodegenerative disorder that is part of the so-called spongiform encephalopathies, which is caused by the abnormal accumulation of PrP protein (called PrPSc) in the central nervous system. The most common human prion disease is sporadic form of Creutzfeldt-Jakob, whose appearance has been associated with environmental effects or unknown and random genetic events that result in the spontaneous production of PrP in the brain.
In this work we will present two Clinical cases of two woman who visited the emergency room of the hospital Universitario San Ignacio, in which a rapidly progressive encephalopathy caused by Creutzfeldt-Jakob disease is suspected.
Key words: Creutzfeld-Jakob disease, 14-3-3 protein, Tau protein, rapidly progressive dementia, RMN DWI (diffusion weighted imaging) (MeSH).
Introducción
La enfermedad priónica (EP) en humanos hace parte del grupo de las encefalopatías espongiformes transmisibles, son un conjunto de enfermedades neurodegenerativas cuyo agente responsable es una isoforma anormal de una proteína celular denominada proteína priónica, la cual sufre una alteración en su estructura secundaria en un proceso de postraducción generando partículas infecciosas1.
Existen tres formas de enfermedades priónicas: hereditaria, adquirida o esporádica. Esta última, de acuerdo a sus características clínicas y patológicas, se divide en Creutzfeld Jakob (ECJ), insomnio familiar fatal y el Kuru2 (tabla 1).
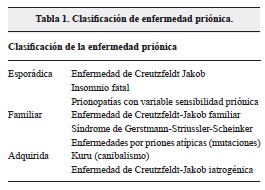
La enfermedad de Creutzfeld-Jakob representa más del 90 % de todos los casos de prionopatía esporádica y puede ser categorizada en cuatro subtipos que se distinguen de acuerdo con los signos clínicos, lesiones histológicas y características moleculares de la proteína priónica patogénica (PRPsc).
Este desorden neurodegenerativo es rápidamente progresivo y fatal entre los 6-8 meses después de su instauración, y la edad promedio de presentación es 65 años (rango entre 30-90 años)3.
El diagnóstico preciso de la enfermedad priónica depende de un cuidadoso examen clínico y de la interpretación de pruebas diagnósticas, que incluyen electroencefalograma, marcadores como la proteína 14-3-3, proteína Tau y neuroimágenes4.
Reportamos dos casos de pacientes femeninas que consultan por cuadro clínico de demencia rápidamente progresiva, en quienes los estudios paraclínicos e imaginológicos realizados sugieren diagnóstico de enfermedad priónica.
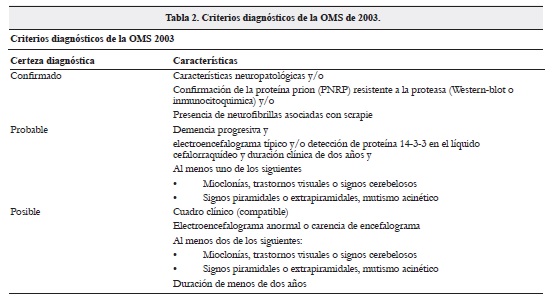
Criterios diagnósticos
En los últimos años se han propuesto múltiples criterios diagnósticos que contemplan dentro un contexto clínico adecuado el uso de pruebas diagnósticas, como se expone en las tablas 2 y 3.

Presentación del caso 1
Paciente femenina de 63 años de edad, sin antecedentes patológicos, quien ingresó al servicio de urgencias por cuadro clínico de tres semanas de evolución se exacerbó durante los últimos cinco días, consistente en sensación rotacional externa que se incrementaba con los cambios de posición y mejoraba con el reposo, asociado a bradilalia, bradipsiquia, episodios de desorientación e insomnio de conciliación. Consultó previamente otra institución, donde le realizaron Resonancia Magnética Nuclear (RMN ) de cerebro simple reportada como normal. Al examen neurológico se documentó compromiso cerebeloso, de memoria, de cálculo y del lenguaje con conductas utilitarias, por lo cual se consideró que inicialmente cursó con encefalopatía aguda y diagnósticos diferenciales de encefalitis de Hashimoto, paraneoplásica vs. encefalitis mediada inmunológicamente versus menos probablemente prionopatía esporádica.
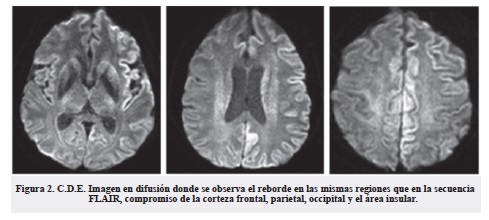
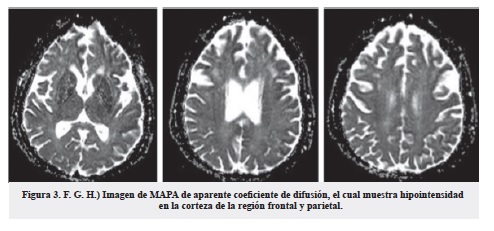
Se inició estudio intrahospitalario, por lo cual se solicitó TSH y T4 normales, punción lumbar con presión de apertura normal y citoquímico con leve hiperproteinorraquia; resonancia magnética cerebral simple que reportó lesiones hiperintensas inespecíficas en la sustancia blanca supratentorial, probablemente secundarias a microangiopatía isquémica crónica o hipertensiva (Fazekas II) con leve atrofia cerebral cortical esperada para la edad; y electroencefalograma que evidenció compromiso encefalopático con actividad asilada pseudoperiódica en la región temporal izquierda (figuras 1-4).
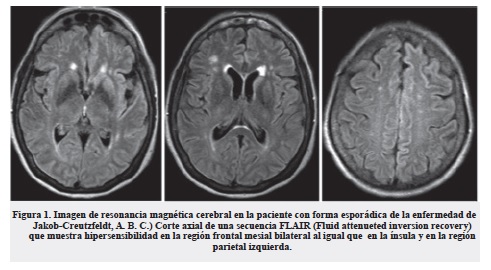
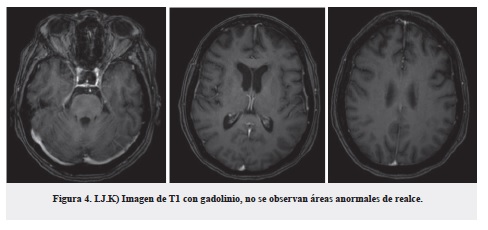
Durante la estancia hospitalaria presentó rápido deterioro de la conciencia hasta llegar al estupor, así como múltiples episodios psicóticos, por lo cual se realizó segunda RMN de cerebro simple y con gadolinio con evidencia de restricción de señal en la corteza, predominantemente del hemisferio cerebral izquierdo, evidentes en las secuencias de difusión y nuevo electroencefalograma en el que se documentan ondas periódicas tipo PLEDs en la región temporal izquierda, que se irradian a la región frontal ipsilateral y en ocasiones toman carácter de BIPLEDs, indicando compromiso encefalítico con posibilidad de prionopatía.
Como manejo médico la paciente recibió cinco bolos de metilpredinosolona, ácido valproico durante 20 días, y tres sesiones de plasmaferesis sin respuesta alguna a las acciones terapéuticas instauradas.
En vista de la pobre respuesta al manejo farmacológico y el empeoramiento del estado general, se da egreso hospitalario un mes después con control ambulatorio por neurología.
Presentación del caso 2
Paciente femenina de 62 años de edad, sin antecedentes patológicos, quien asiste a servicio de urgencias del Hospital Universitario San Ignacio por cuadro clínico de dos meses de evolución, consistente en fallas en la memoria episódica, semántica, procedimental y de trabajo. Al examen neurológico se encontró a la paciente alerta, hipoproséxica, orientada en persona y lugar, pero desorientada en tiempo.
Lenguaje con bloqueos, intrusiones, ocasionalmente con anomia en su discurso y escritura con evidencia de agramatismos. No logró realizar abstracción ni conteo regresivo. El resto del examen neurológico sin alteraciones.
La paciente llevó las pruebas neuropsicológicas extrainstitucionales realizadas en enero de 2015, que mostraron deterioro cognitivo de posible etiología vascular, con funcionamiento cognoscitivo disminuido, afasia semántica, compromiso de memoria verbal explícita y dificultades de tipo atencional a nivel ejecutivo. Se consideró, por tanto, que la paciente cursó con un cuadro de demencia rápidamente progresiva y se inició estudio intrahospitalario mediante punción lumbar con presión de apertura normal y citoquímico sin evidencia ningún hallazgo patológico; TAC de abdomen y de tórax sin alteraciones; electroencefalograma que mostró anomalías dadas por ondas lentas que tienden a tomar un patrón pseudoperiódico de predominio en región frontotemporal derecha; y el RM simple y con gadolinio evidenció restricción de la difusión que compromete la corteza cerebral de los lóbulos frontales, parietales, occipitales y temporales de manera bilateral y simétrica.
Se inició manejo empírico con bolos de metilprednisolona (recibió 3 dosis en total), sin mejoría de la sintomatología, por lo cual la paciente fue dada de alta con orden de control ambulatorio por parte del servicio de neurología.
Discusión
La enfermedad de Cretuzfeldt-Jakob pertenece a una pequeña, rara y fatal familia de desórdenes degenerativos, que se relaciona con una alteración de una proteína priónica intracelular (PrP).
Dada la morbimortalidad asociada a esta patología y al alto riesgo de transmisión, el diagnóstico oportuno de esta enfermedad es vital. Sin embargo, actualmente los criterios son controversiales y su diagnóstico se hace usualmente en las fases terminales.
Este artículo tiene como finalidad realizar una revisión detallada de la literatura, nos enfocamos en los métodos diagnósticos disponibles en nuestro medio, y su capacidad de permitir la diferenciación con otras demencias rápidamente progresivas, que cursan con manifestaciones clínicas similares.
Ya que el diagnostico definitivo de la enfermedad requiere de confirmación neuropatológica, usualmente post mórtem, se debe tomar en cuenta los signos clínicos como ataxia cerebelosa, demencia rápidamente progresiva y mioclonos, además de una serie de estudios claves como resonancia magnética, análisis del líquido cefalorraquídeo y electroencefalograma para llegar a un diagnóstico temprano y oportuno3.
Los marcadores obtenidos del LCR como la proteína 14-3-3, Tau y enolasa específica de neuronas, son inespecíficos de injuria, muerte neuronal rápida y activación de células gliales, por lo que en varios estudios se han intentado combinar para hacer más preciso el diagnóstico.
Recientemente Forner et, dada la pobre sensibilidad y especificidad de CSF 14-3-3 para la Enfermedad de Creutzfeldt-Jakob esporádica (sCJD) en la evaluación de los pacientes con demencia rápidamente progresiva (RPD) y la discordancia entre los diferentes marcadores de LCR utilizados para el diagnostico de esta entidad, realizaron una evaluación donde compararon 14-3-3, ESN, y T-tau en una cohorte de pacientes remitidos a la Universidad de California en San Francisco (UCSF) y el Centro de Memoria y Envejecimiento entre el 2005 y el 2012, en la tabla 4 se muestran los resultados obtenidos.
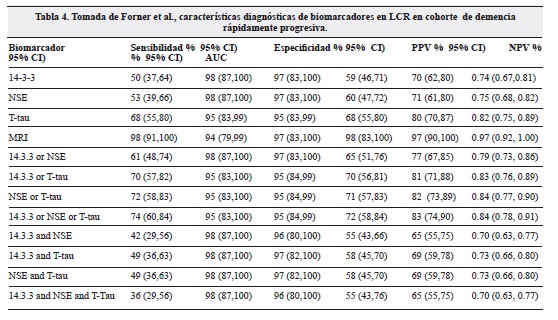
Además Forner et al., también evaluó la relación entre estos tres marcadores y la RMN DWI (diffusion weighted imaging), encontrando ésta última como mejor predictora de ECJ con AUC (area under the receiver operator curve) de 0.97 (IC 0.92-1.00) y una precisión diagnostica de 97 % (IC 97-100 %), por lo cual se concluyó que la RM tenía mucha mejor utilidad diagnóstica que cualquier biomarcador de líquido cefalorraquídeo o combinación de los mismos.
Una de las razones por las cuales la 14-3-3, NSE, y T-tau no son tan útiles como la RM, es que son marcadores no específicos de lesión neuronal rápida.
En la cohorte del estudio en mención, T-tau era una mejor medida de diagnóstico de sCJD, comparada con 14-3-3 o NSE, en parte por su mayor sensibilidad y ausencia de falsos positivos, siendo sin embargo, mucho menos precisa que la RM5,6 .
Presentación de resultados
Se enviaron las muestras de LCR de las dos pacientes al Centro de Vigilancia Nacional de Enfermedad Priónica en Estados Unidos. Respecto a la primera paciente, encuentraron la proteína Tau positiva con valor de 3120pg/ml (punto de corte 1150 pg/ml) y proteína 14-3-3 positiva. En contraste, la segunda paciente presenta proteína Tau negativa con valor de 373 pg/ml (punto de corte: 1150 pg/ml) mientras que la proteína 14-3-3 fue positiva. Lo último podría atribuirse a que los niveles de la proteína Tau en LCR suelen estar en bajos niveles dos meses antes de la instauración del cuadro y aumentar a las seis semanas del mismo6-10.
Es fundamental recordar que, como vimos en las imágenes anteriores, ambas pacientes presentaron imágenes altamente sugestivas de enfermedad priónica.
Se concluye que según los criterios diagnósticos de la OMS y la Universidad de San Francisco (USF), ambas pacientes tendrían enfermedad de Creutzfeldt-Jakob probable.
Conclusiones
Los datos presentados sugieren que la adquisición de resonancia magnética del cerebro, incluida la recuperación de inversión atenuada por líquido, DWI y las secuencias de coeficiente de difusión aparente, debe ser uno de los pilares de la evaluación de los pacientes con sospecha de sCJD, particularmente en los casos atípicos o cuando las pruebas de LCR muestren resultados contradictorios o negativos.
Conflicto de intereses
Los autores declaran no presentar conflicto de intereses.
Referencias
1. RODRIGUEZ DR, CILLIANI B. Encefalopatías espongiformes transmisibles. Infectio. 2004;8:9. [ Links ]
2. Puoti G, Bizzi A, Forloni G, Safar JG, Tagliavini F, Gambetti P. Sporadic human prion diseases: Molecular insights and diagnosis. Lancet Neurol. Elsevier Ltd; 2012;11(7):618-28. [ Links ]
3. VAN EVERBROECK B, BOONS J, CRAS P. Cerebrospinal fluid biomarkers in Creutzfeldt-Jakob disease. Clin Neurol Neurosurg. 2005;107(5):355-60. [ Links ]
4. BARASHI NS, VARGAS C, ZARCO LA. Enfermedades priónicas humanas. Univ Medica. 2013; 54(4):495-516 [ Links ]
5. FORNER SA, TAKADA LT. Comparing CSF biomarkers and brain MRI in the diagnosis of sporadic Creutzfeldt-Jakob disease. Neurology Clinical practice 2015,116-125. [ Links ]
6. COULTHART, MB, ANCES BM. Creutzfeldt-Jakob disease the search for definitive diagnostic tests continues. Neurology Clinical practice 2015, 99-101. [ Links ]
7. SHINOHARA MN, HAMAGUCHI T. Serum tau protein as a marker for the diagnosis of Creutzfeldt-Jakob disease. J Neurol 2011, 258, 1464-1468. [ Links ]
8. ZANUSSO G, FIORINI M, FERRARI S, ET AL. Cerebrospinal Fluid Markers in Sporadic Creutzfeldt-Jakob Disease. International Journal of Molecular Sciences. 2011;12(9):6281-6292. [ Links ]
9. COULTHART MB, JANSEN GH, OLSEN E, ET AL. Diagnostic accuracy of cerebrospinal fluid protein markers for sporadic Creutzfeldt-Jakob disease in Canada: a 6-year prospective study. BMC Neurology. 2011;11:133. [ Links ]
10. KARCH A, HERMANN P, PONTO C ET AL. Cerebrospinal fluid tau levels are a marker for molecular subtype in sporadic Creutzfeldt-Jakob disease. Neurobiology of aging 2015,1964-1968. [ Links ]

