










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.32 no.3 Bogotá July/Sept. 2016
Caso Clínico
Enfermedad de Pompe como diagnóstico diferencial de enfermedad de motoneurona: reporte de casos y revisión de la literatura
Pompe disease as a differential diagnosis of motor neuron disease: report of a case and review of the literature
Lina M. Ariza-Serrano (1), Jesús Hernán Rodríguez-Quintana (2), Rafael Serrano (3), Rogelio Camacho (4)
(1) Residente de Neurología, Universidad del Rosario, Fundación Cardioinfantil, Bogotá, Colombia
(2) Jefe de posgrados en Neurología, neurólogo, neurofisiólogo, Universidad del Rosario, Fundación Cardioinfantil, Bogotá, Colombia
(3) Neurólogo, Clínica VIP, Bogotá, Colombia
(4) Fisiatra, Fundación Cardioinfantil, Bogotá, Colombia
Recibido: 12/04/16. Aceptado: 25/05/16.
Correspondencia: Lina M Ariza-Serrano, ariza.linamaria@gmail.com
Resumen
A través del presente estudio se pretende demostrar la importancia de la evaluación de la enfermedad de Pompe como diagnóstico diferencial de la enfermedad de motoneurona. En el siguiente trabajo presentamos dos casos clínicos en los que inicialmente se consideró enfermedad de motoneurona, y en donde finalmente se documentó un déficit de alfa glucosidasa como causal de la sintomatología.
Palabras clave: Pompe, Motoneurona, Diagnóstico diferencial (DeCS).
Summary
Through this study we aim to demonstrate the importance of the evaluation of pompe disease as a differential diagnosis of motor neuron disease. Here we present two cases in which the initial approach was of a motor neuron disease, but with a more comprehensive assessment it was documented an alpha glucosidase deficiency.
Key words: Pompe, Motor neuron disease, Differential diagnosis(MeSH).
Introducción
Las enfermedades de motoneurona se pueden clasificar según su compromiso en enfermedad motoneurona superior o inferior y según su tiempo de evolución, de inicio agudo o inicio crónico. La enfermedad de Pompe puede cursar con características clínicas muy similares a la presentación de estas enfermedades y es un diagnóstico diferencial a tener en cuenta en estos pacientes. Se cree que del 5 al 8 % de los pacientes con esclerosis lateral amiotrófica (ELA), tienen un diagnóstico alternativo, y hasta un 50 % de estos pacientes puede ser tratado. La Federación Europea de Sociedades Neurológicas (EFNS; por sus siglas en inglés) clasifica la enfermedad de Pompe como un diagnóstico diferencial de la ELAi. A continuación presentamos dos casos, uno de ellos diagnosticado inicialmente como enfermedad de motoneurona, y el otro como miopatía, en los que posterior a los estudios realizados se encontró un déficit de alfa glucosidasa como causal de la sintomatología del paciente.
Presentación de los casos
Caso 1: paciente de 34 años, que consultó en el año 2006 con cuadro clínico de tres años de evolución de debilidad generalizada con predominio proximal, atrofia muscular y arreflexia (Figuras 1 y 2). Se le realizó CPK total, la cual estuvo elevada (643 UI/L), y electromiografía + neuro-conducción (EMG + NC) de las cuatro extremidades que mostraron valores de amplitud, latencias y velocidad de conducción dentro de la normalidad. También se estudió con una Resonancia Magnética Nuclear (RMN) lumbar que mostró una discopatía L5-S1 con protrusión discal que no era compresiva. Ante la alta sospecha de miopatía se realizó una biopsia muscular que no resultó conclusiva. En el 2009 el paciente inició con disnea progresiva, tos persistente con expectoración purulenta, asociada a síntomas constitucionales, que se relacionaron con debilidad en los músculos respiratorios con sobreinfección pulmonar, lo que lo llevó a intubación oro-traqueal (IOT) y Bilevel positive airpressure (BPAP) de manera ambulatoria por tienda de traqueostomía de forma permanente. Dos años más tarde, se realizó una nueva EMG + NC que evidenció potenciales de denerva-ción aguda en miotomas de L4, paravertebrales izquierdos y paravertebrales C6 y C7 derechos, además de unidades motoras polifásicas con baja amplitud de los potenciales de acción en los miembros inferiores, y ausencia de onda F en los nervios peroneo y tibial bilateral. Ante la sospecha de que se tratara de una enfermedad de motoneurona de tipo atrofia muscular espinal, se iniciaron terapias de rehabilitación en casa y se realizó reacción en cadena de la polimerasa (PCR) para atrofia muscular espinal (SMN1), la cual resultó negativa. En el 2013, por aumento en la dificultad respiratoria se le implantó un marcapasos diafragmático. En ese año, se le realizó prueba de filtro para la enfermedad de Pompe, en búsqueda de diagnósticos diferenciales, se encontró que estaba dentro de los límites normales. Sin embargo, dada la clínica que sugería esta enfermedad se solicitó la prueba de leucocitos, que determinó un probable déficit de alfa glucosidasa. Posteriormente, se realizó el test de secuencia-ción genética GAA que confirmó la enfermedad de Pompe, con dos mutaciones GAA heterocigotas. Con las pruebas confirmatorias de la enfermedad, se inició manejo con alfa glucosidasa. Actualmente el paciente se encuentra en suplencia enzimática, continúa cuadriparético, con atrofia muscular generalizada y debilidad de predominio proximal, con requerimiento de BPAP y cánula de traqueostomía.
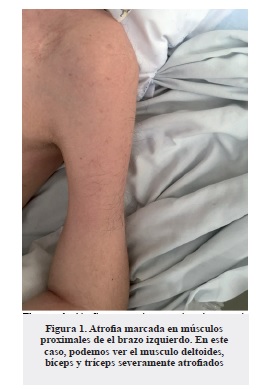
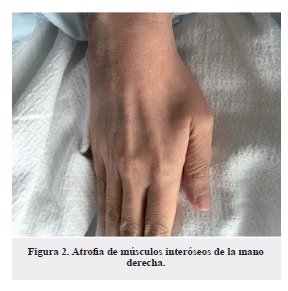
Caso 2: paciente de 60 años, que presentó un cuadro clínico de diez años de evolución de debilidad proximal y progresiva en miembros inferiores, lo que dificultó la marcha, especialmente para subir escaleras, y se asoció a dolor. Concomitantemente, el paciente presentó dolor braquial bilateral, de predominio proximal que aumentaba al realizar actividad física, con leve hipotrofia muscular asociada. Se le realizó CPK (427 U/L), LDH (373 UI/L), Anti-Ro positivo (50,3), con el resto de ENAS negativos. Se le realizó EMG+NC que muestran potenciales de unidad motora de características miopáticas (baja amplitud, corta duración y polifásicas) en músculo trapecio, deltoides derecho, tensor de la facia alta bilateral y peroneos). La biopsia muscular mostró un proceso miopático severo (secundario a distrofia, miopatía vacuolar por enfermedad de depósito o miositis por cuerpos de inclusión). Fue valorado inicialmente por reumatología, quienes ante la sospecha de que se tratase de una polimiositis iniciaron manejo con metrotexate semanal, y posteriormente con azatioprina sin adecuada respuesta. El paciente fue reenviado a neurología para segundo concepto. Se descartó presencia la de enfermedad de motoneurona y se le realizó la prueba de filtro para la enfermedad de Pompe en búsqueda de diagnósticos diferenciales. El resultado de este test sugiere la presencia de la enfermedad, por lo que se realizó la secuenciación genética, que confirmó la enfermedad de Pompe, con dos mutaciones GAA heterocigotas. Se inició suplencia enzimática, con mejoría de los síntomas previamente descritos. El paciente actualmente es totalmente funcional en todas las actividades de su vida diaria.
Revisión del la literatura
La enfermedad de motoneurona, de tipo superior o inferior, incluye las células de Betz en la corteza motora, y la neurona motora inferior, ya sea en los núcleos del tallo cerebral o en el asta anterior de la médula espinal2,3. La esclerosis lateral amiotrófica (ELA), es considerada la enfermedad de motoneurona más frecuente, teniendo en América Latina una incidencia anual aproximada de 0.2 casos por 100.000 habitantes en Ecuador, 0.7 casos por 100.000 habitantes en Costa Rica, 0.4 casos por 100.000 habitantes en Brasil, 1.37 casos por 100.000 habitantes en Uruguay y 3.17 casos por 100.000 habitantes en Argentina4. Afecta con mayor frecuencia a los hombres, y aproximadamente el 5 % de los casos tienen una factor familiar implicado5,6. Aunque clásicamente esta enfermedad se caracteriza por su compromiso motor, tanto de motoneurona inferior como superior, en la actualidad es evidente que tiene un compromiso multisisté-mico, extrapiramidal y cognitivo5-7. Para su diagnóstico, se usan mundialmente los criterios del escorial, los cuales permiten realizar un diagnóstico definitivo, probable o posible6. La guía de manejo de la Federación Europea de Sociedades Neurológicas recomienda la realización de análisis de ADN para genes SOD1, SMN, SBMA, TDP43 y FUS, en casos seleccionados y como nivel del evidencia clase IV1, debido a que las personas con una de estas mutaciones y la presencia de signos clínicos de compromiso de motoneurona superior y/o inferior tendrían una ELA clínicamente definitiva1.
Del 1 al 4 % de las enfermedades de motoneurona cursan con compromiso superior único, y con un curso clínico más benigno, que se denomina esclerosis lateral primaria (ELP)8. La ELA y paraplejía espástica hereditaria son los diagnósticos diferenciales de principal consideración8. La ELP, al igual que la ELA, es más frecuente en hombres8. Para su diagnóstico certero, se requiere la ausencia de fasci-culaciones o atrofia, alteraciones sensitivas al examen físico o historia familiar de una enfermedad similar8.
La atrofia muscular espinal (AMS) afecta el asta anterior de la médula espinal, lo que lleva a la aparición de atrofia y debilidad muscular asimétrica de predominio proximal2, en la que se estima una incidencia promedio de 1 en 11.000 nacidos vivos9. Su forma de presentación más común se relacionada con la mutación del gen SMNi, la cual se hereda de manera autosómica recesivai0. Clínicamente la AMS se clasifica según el grado de compromiso motor y la edad de presentación de la misma10. La tipo 3 y 4 se presentan en la edad adulta, siendo la tipo 4, la más benigna de todas las AMS10. La atrofia muscular progresiva (AMP), con compromiso de motoneurona inferior aislado, y de fisio-patología aún incierta, cursa con atrofia, fasciculaciones e hiporeflexia; y tiene una incidencia aproximada de 0.02 por cada 100.00011,12. Es más frecuente en hombres y tiene un mejor pronóstico que el de los pacientes con ELA12. La edad de aparición es más tardía que en la ELA, con un promedio de inicio de 63 años12. Es posible que los pacientes inicialmente diagnosticados como AMP, con el paso del tiempo manifiesten signos de compromiso de motoneurona superior y deban ser reclasificados a una ELA12.
Las ganglionopatías con compromiso sensitivo puro se presentan cuando hay una lesión localizada en el ganglio de la raíz dorsal13,14. El patrón de presentación es asimétrico con pérdida sensorial con compromiso propioceptivo, con ataxia temprana y arreflexia15,13. La teoría actual de esta injuria se debe a una reacción cruzada, mediada por células T citotóxicas (Cd8+) entre las proteínas neuronales expresadas en el cáncer y los antígenos neuronales14. La mayor asociación que se ha encontrado hasta el momento es con la proteína de unión HuD (ELAVL3) que es expresada tanto en el SNC, como el cáncer de células pequeñas de pulmón14.
En cuanto a la enfermedad de Pompe, aún considerada una enfermedad rara, es una glucogenosis, autosómica recesiva, en donde se producen acúmulos de glicógeno intralisosomal a causa del déficit parcial o total en la enzima alfa glucosidasa ácida, secundaria a mutaciones en el gen GAA16. La frecuencia aproximada de esta enfermedad es de 1 por cada 40.000 personas, y las frecuencias más altas se presentan en Taiwan y en Estados Unidos17. Se clasifica comúnmente de acuerdo a su edad de inicio en aquellas de la infancia (IOPD - infantile onsetpompe disease), y aquellas de inicio más tardío (LOPD - Late onset Pompe disease)16. En la infancia, se manifiesta con debilidad e hipotonía que se asocia a hepatomegalia, cardiomiopatías y macroglosia secundaria a los acúmulos de glicógeno en estos órganos. En el adulto, se asocia a anomalías de la conducción cardiaca, como por ejemplo el Wolff Parkinson White (WPW), insuficiencia respiratoria secundaria a acúmulos en los músculos intercostales y diafragma, miopatía progresiva con compromiso apendicular predominante, debilidad en la lengua (depósitos en el geniogloso) y ptosis uni o bilate-ral16. El diagnóstico es clínico, histológico (glicógeno unido a la membrana citoplasmática intra-miofibrilar, vacuolas de glicógeno visibles con tinción de PAS (Periodic acid-schift) en biopsia de músculo, por medición de la actividad enzimática en sangre (usada normalmente como tamizaje), que puede ser confirmada mirando la función enzimática en algún otro tejido o realizando la secuenciación genética de el gen GAA16. El tratamiento con suplencia enzimática se encuentra disponible desde hace años, y ha demostrado un crucial beneficio, sobre todo en estadios iniciales de la enfermedad18. Aumenta la supervivencia, reduce el riesgo de ventilación mecánica y mejora la fuerza muscular esquelética y respiratoria18.
Los casos clínicos reportados hasta el momento muestran que la clínica presentada por estos pacientes se puede llegar a correlacionar con el tejido afectado en autopsia, por ejemplo en compromiso proximal del músculo esquelético de las extremidades y debilidad. Sin embargo, hay otros tejidos en los que no se ha encontrado una asociación patológico - clínica, como es el caso de la gliosis fibrilar, la degeneración de las células del asta anterior de la médula, la acumulación vacuolar de glicógeno en las células de Schwann en los nervios periféricos, entre otros16. Hasta donde llega nuestro conocimiento, consideramos importante que se investigue esta enfermedad en el adulto, ya que aunque es una enfermedad rara, tiene el beneficio de tener un tratamiento, a través de la suplencia enzimática, que mejore la calidad de vida, reduzca complicaciones y aumente la supervivencia de estos pacientes.
Conflicto de intereses
Los autores manifiestan no tener conflictos de intereses en este estudio.
Referencias
1. Andersen PM, Abrahams S, Borasio GD, de Carvalho M, Chio A, et al. EFNS guidelines on the Clinical Management of Amyotrophic Lateral Sclerosis (MALS) - revised report of an EFNS task force. Eur J Neurol [Internet]. Blackwell Publishing Ltd; 2012 Mar 1;19(3):360-75. Available from: http://dx.doi.org/10.1111/j.1468-1331.2011.03501.x. [ Links ]
2. Statland JM, Barohn RJ, McVey AL, Katz JS, Dimachkie MM. Patterns of Weakness, Classification of Motor Neuron Disease, and Clinical Diagnosis of Sporadic Amyotrophic Lateral Sclerosis. Neurol Clin [Internet]. 2015 Nov [cited 2015 Nov 15];33(4):735-48. Available from: http://www.sciencedirect.com/science/article/pii/S0733861915000638. [ Links ]
3. Báumer D, Talbot K, Turner MR. Advances in motor neurone disease. J R Soc Med [Internet]. 2014;107(1):14-21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24399773. [ Links ]
4. Zapata-Zapata C, Franco-Dager E, Solano-Atehortúa J, Ahunca-Velásquez L. Esclerosis lateral amiotrófica: Actualización. IATREIA. 2016;29(2):194-205. [ Links ]
5. Linden-Junior E, Becker J, Schestatsky P, Rotta FT, Marrone CD, Gomes I. Prevalence of amyotrophic lateral sclerosis in the city of Porto Alegre, in Southern Brazil. Arq Neuropsiquiatr. 2013;71:959-62. [ Links ]
6. Harms MB, Baloh RH. Clinical Neurogenetics: Amyotrophic Lateral Sclerosis. Neurol Clin. 2013;31(4):929-50. [ Links ]
7. Ingre C, Roos PM, Piehl F, Kamel F, Fang F. Risk factors for amyotrophic lateral sclerosis. Clin Epidemiol [Internet]. 2015;7,181-93. Available from: http://www.dovepress.com/risk-factors-for-amyotrophic-lateral-sclerosis-peer-reviewed-fulltext-article-CLEP. [ Links ]
8. Statland JM, Barohn RJ, Dimachkie MM, Floeter MK, Mit-sumoto H. Primary Lateral Sclerosis. Neurol Clin [Internet]. 2015 Nov [cited 2015 Nov 22];33(4):749-60. Available from: http://www.sciencedirect.com/science/article/pii/S073386191500064X. [ Links ]
9. Sugarman EA, Nagan N, Zhu H, Akmaev VR, Zhou Z, Rohlfs EM, et al. Pan-ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: clinical laboratory analysis of 72[thinsp]400 specimens. Eur J Hum Genet [Internet]. Macmillan Publishers Limited; 2012 Jan;20(1):27-32. Available from: http://dx.doi.org/10.1038/ejhg.2011.134. [ Links ]
10. Kolb SJ, Kissel JT. Spinal Muscular Atrophy. Neurol Clin [Internet]. 2015 Nov [cited 2015 Nov 22];33(4):831-46. Available from: http://www.sciencedirect.com/science/article/pii/S0733861915000614. [ Links ]
11. Kim W-K, Liu X, Sandner J, Pasmantier M, Andrews J, Rowland LP, et al. Study of 962 patients indicates progressive muscular atrophy is a form of ALS. Neurology [Internet]. American Academy of Neurology; 2009 Nov 17;73(20):1686-92. Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2788803/. [ Links ]
12. Liewluck T, Saperstein DS. Progressive Muscular Atrophy. Neurol Clin [Internet]. 2015 Nov [cited 2015 Nov 18];33(4):761-73. Available from: http://www.sciencedirect.com/science/article/pii/S0733861915000626. [ Links ]
13. Barohn RJ, Amato AA. Pattern-recognition approach to neuropathy and neuronopathy. Neurol Clin [Internet]. 2013 May [cited 2015 Nov 18];31(2):343-61. Available from: http://wwwsciencedirect.com/science/article/pii/S073386191300011X. [ Links ]
14. Gwathmey KG. Sensory Neuronopathies. Muscle Nerve [Internet]. 2015 Oct 1;n/a - n/a. Available from: http://dx.doi.org/10.1002/mus.24943. [ Links ]
15. Zuberbuhler P, Young P, León Cejas L V, Finn BC, Bruetman JE, Calandra CR, et al. [Sensory neuronopathy. Its recognition and early treatment]. Medicina (B Aires) [Internet]. Servicio de Neurología Hospital Teodoro álvarez, Buenos Aires, Argentina. Fundación Revista Medicina (Buenos Aires); 2015;75(5):297-302. Available from: http://search.ebscohost.com/login.aspx?direct=true&db=mdc&AN=26502464&lang=es&site=ehost-live. [ Links ]
16. Dasouki M, Jawdat O, Almadhoun O, Pasnoor M, McVey AL, Abuzinadah A, et al. Pompe disease: Literature review and case series. Neurologic Clinics. 2014. p. 751-76. [ Links ]
17. Lim J-A, Li L, Raben N. Pompe disease: from pathophysiology to therapy and back again. Front Aging Neurosci [Internet]. 2014;6:177. Available from: http://journal.frontiersin.org/article/10.3389/fnagi.2014.00177/abstract. [ Links ]
18. Mellies U, Lofaso F. Pompe disease: A neuromuscular disease with respiratory muscle involvement. Respiratory Medicine.2009. p. 477-84. [ Links ]

