










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.32 no.4 Bogotá Oct./Dec. 2016
https://doi.org/10.22379/24224022113
https://doi.org/10.22379/24224022113
Caso clínico
Enfermedad de moyamoya: a propósito de un caso
Moyamoya disease: a case report
Ledmar Jovanny Vargas R (1), Sergio José Sánchez H (1), Álvaro Suárez Chaparro (2), Oscar M. Jiménez Peña (3)
(1) Estudiante Medicina, décimo semestre, Universidad de Boyacá, Tunja, Colombia
(2) Neurocirujano y cirujano de columna, Magister en Salud Pública, Jefe del Servicio de Neurocirugía, Hospital San Rafael, docente Universidad de Boyacá, Tunja, Colombia
(3) Odontólogo Ph.D., jefe Departamento Salud Pública, docente tiempo completo, Universidad de Boyacá, Tunja, Colombia
Recibido: 12/08/15. Aceptado: 13/05/16.
Correspondencia: Ledmar Vargas, lejovaro@gmail.com
Resumen
La enfermedad de moyamoya (EMM) es una patología crónica caracterizada por la oclusión progresiva de la vasculatura cerebral y no aterosclerótica, generando un patrón angiográfico conocido como moyamoya, que en japonés hace alusión a la apariencia en "bocanada de humo", trae como consecuencia eventos cerebrovasculares isquémicos o hemorrágicos. Tiene dos picos de presentación, uno entre los 5 y 9 años y otro entre los 45 y 49 años, con un ligero predominio en mujeres, suele ser más frecuente en individuos de origen asiático, y menos en personas de origen hispano. El tratamiento de elección es el procedimiento quirúrgico precoz y su pronóstico no es totalmente predecible.
Reporte de caso: mujer de 44 años que ingresó por presentar movimiento tónico-clónicos y requirió IOT, con tomografía axial computarizada cerebral simple que mostró hematoma intraparenquimatoso izquierdo y arteriografía con patrón moyamoya. Se manejó en la unidad de Cuidados Intensivos, donde finalmente falleció.
Palabras clave: enfermedad cerebrovascular, moyamoya, hemorragia intracerebral, artereopatía oclusiva intracraneal progresiva (DeCS).
Summary
Moyamoya disease (EMM) is a chronic disease characterized by progressive occlusion of the cerebral vasculature and nonatherosclerotic, generating an angiographic pattern known as moyamoya, which in Japanese refers to the appearance "puff of smoke", results in cerebrovascular ischemic or hemorrhagic events. Presentation has two peaks, one between 5 and 9 years and another between 45 and 49 years, with a slight predominance in women over men, usually more common in individuals of Asian origin, and less in people of Hispanic origin. The treatment of choice is early surgical procedure and prognosis is not entirely predictable.
Case report: 44-year-old woman presenting income tonic-clonic movement and required IOT, single computed tomography showed left cerebral intracerebral hematoma and arteriography with Moyamoya pattern. It was handled in Intensive Care Unit, where he eventually died.
Key words: cerebrovascular disease, moyamoya, intracerebral hemorrhage, intracranial artery occlusive progressive (MeSH).
Introducción
La enfermedad de moyamoya (EMM) es una patología crónica caracterizada por la oclusión progresiva de la vasculatura cerebral, no aterosclerótica, descrita por primera vez en 1957 por Takeuchi y Shimizu1, con compromiso particular del polígono de Willis1, en la porción terminal de las arterias carótidas internas2 y regiones proximales de las arterias cerebrales anteriores y medias3, que trae como consecuencia dilatación de los pequenños vasos adyacentes, con el fin de convertirse en vías de circulación sanguínea colateral3, generando un patrón angiográfico conocido como moyamoya, nombre dado en 1969 por Suzuki y Takaku, que en japonés hace alusión a la apariencia en "bocanada de humo" de la red arterias colaterales en las imágenes angiográficas4. Esto trae como consecuencia eventos cerebrovasculares isquémicos o hemorrágicos, debido a la oclusión de los vasos del polígono de Willis o a la ruptura por dilatación excesiva de los pequeños vasos adyacentes1.
Esta es una enfermedad huérfana (definida como aquella patología crónicamente debilitante, grave, que amenaza la vida y con una prevalencia menor de 1 por cada 5.000 personas), en Colombia están contempladas en la Resolución 2048 del 20155. Esta enfermedad tiene dos picos de presentación, uno entre los 5 y 9 años y otro entre los 45 y 49 años, con un ligero predominio en mujeres sobre hombres6,7.
La etiología aun es desconocida, aunque algunos estudios refieren que puede deberse a factores genéticos, de hecho se ha relacionado con alteraciones de los cromosomas 3, 6 y 17, además a factores ambientales, como la radioterapia en procesos intracraneanos que incrementan el riesgo de aparición2,7. Existe una forma familiar de la enfermedad que tiene un patrón de herencia autosómico dominante con penetrancia completa y da cuenta del 15 % de los casos6,7.
En los estudios histológicos se han encontrado varias alteraciones estructurales en las capas de las arterias carótidas: engrosamiento fibrocelular de la capa íntima que es secundario a proliferación de células de músculo liso; irregularidades de la lámina elástica interna y adelgazamiento de la túnica media6,7. Por otra parte, se han reportado niveles elevados en el suero y/o líquido cefalorraquídeo de factores de crecimiento y otros péptidos, los cuales contribuirían con el desarrollo de las alteraciones anatomopatológicas propias de la enfermedad6,7.
A continuación presentamos la descripción de una paciente tratada por el servicio de neurocirugía en el Hospital San Rafael de la ciudad de Tunja con diagnóstico confirmado de enfermedad de moyamoya (EMM).
Reporte de caso
Paciente de sexo femenino de 44 años de edad, con antecedentes patológicos de epilepsia tónico clónica (8 años de evolución manejada con fenitoína), accidente cerebro vascular (ACV) isquémico (hace 10 años), resección de quiste mamario, histerectomía abdominal, que es llevada al servicio de urgencias del Hospital Sarare (Arauca) por presentar cuadro clínico caracterizado por movimientos tónico clónicos generalizados con deterioro súbito del nivel de conciencia y sin recuperación de la misma, que duró aproximadamente 30 minutos, motivo por el cual se decidió trasladar a la unidad de cuidados intermedios (UCi), donde requirió con intubación orotraqueal, (IOT y sedación), además se le realizó tomografía computarizada (TC) cerebral que evidenció hematoma intraparenquimatoso temporo-parietal izquierdo (con clasificación de Fisher IV), motivo por el cual se remitió a un hospital de mayor complejidad (Hospital San Rafael de Tunja).
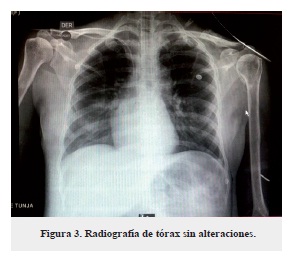
Fue recibida por el servicio de neurocirugía, el cual mencionó en su examen físico que su estado neurológico no era valorable por la sedación. Decidieron iniciar nimodipino, metoclopramida, vasopresores, neuroprotección e ingresarla a la unidad de cuidados intensivos (UCI), además se solicitaron exámenes paraclínicos e instauraron manejo adecuado (tabla 1), y se dejó diagnóstico de lesión supratentorial profunda izquierda subcortical en ínsula.
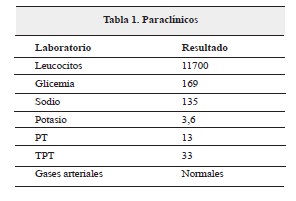
Se realizó panangiografia cerebral, la cual reveló oclusión de arteria carótida interna de forma simetrica, que produce circulación colateral dibujando de forma tenue las arterias cerebral media y anterior, característico del síndrome de moyamoya (figura 1), clasificado en Suzuki III.
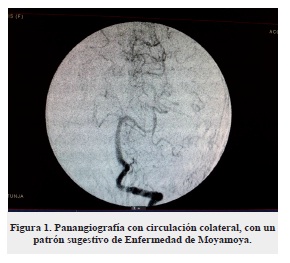
Se realizó segunda TC que reveló hemorragia ganglio-basal izquierda con desviación de la línea media, hipodensidad arteria cerebral media (ACM) derecha más atrofia cerebral del mismo lado, lo que indicó un ACV antiguo (figura 2).
Se propuso realizar revascularización con técnica indirecta en hemisferio cerebral izquierdo, siendo la sinangiosis miocorticodural con injerto pediculado vascular de temporal superficial, el procedimiento de elección; se habló con la hermana (tutora legal en ese momento) quien no aceptó realizar los procedimientos quirúrgicos.
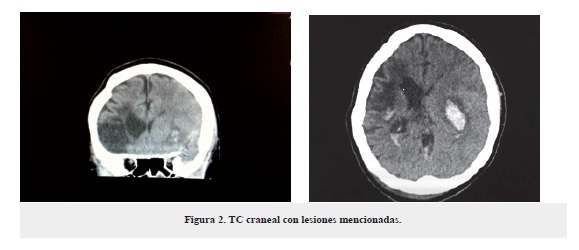
La paciente estuvo varios días (cerca de 6) en la UCI donde presentó alcalosis metabólica (pH 7,51; HCO3 26; CO2 32; SaO2 94; PO2 64), se realizó una radiografía de tórax, la cual no revela ninguna alteración (figura 3).
A los dos días después de retirada la sedación, la paciente presentó postura de decorticación, bradicardia extrema, progresando hacia una asistolia y falleció en horas de la madrugada de su 10° día de hospitalización.
Discusión
Esta entidad suele ser más frecuente en individuos de origen asiático, y menos en personas de origen hispano, como lo demuestran algunos estudios realizados2,8. En Japón se ha encontrado una prevalencia de 3.16 casos por 100.000 habitantes2,9, mientras que en Estados Unidos en un reciente artículo se encontró que la prevalencia de la enfermedad es de 0.086 por cada 100.000 habitantes7.
El caso que presentamos hace referencia a la enfermedad de moyamoya que está ligado a formas idiopáticas de la arteriopatía con compromiso bilateral oclusivo10, a diferencia del síndrome de moyamoya que se refiere a pacientes con lesiones unilaterales asociadas con otras entidades7 como neurofibromatosis, anemia de células falciformes2, drepanocitosis6, síndrome de Down11, meningitis, trauma, enfermedades autoinmunes, secundarias a radiación craneal9 y enfermedad de graves12.
La clínica de esta enfermedad varía con la edad, encontramos que los infantes (5 a 9 años) con moyamoya típicamente se presentan con ictus arteriales isquémicos o accidentes isquémicos transitorios (AIT), mientras que en los adultos (45 a 49 años) son más frecuentes las hemorragias intracraneales13, tal como quedó demostrado en nuestra paciente que tenía los factores sociodemográficos de edad, sexo femenino y además el proceso hemorrágico característico de la edad adulta en esta enfermedad.
Ante la sospecha clínica se deben solicitar estudios radiológicos como tomografía computarizada (TC) de cráneo donde se pueden evidenciar áreas pequeñas de hipodensidad que sugerirían hemorragia o isquemia3, tal como se evidenció en nuestro caso.
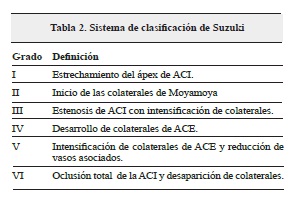
La Imagen por resonancia magnética (IRM) contrastada (con gadolinio) ha mostrado el "signo de la hiedra", el cual es un realce leptomeníngeo difuso que se puede producir en la EMM3,12, aunque no fue posible realizar este estudio por falta de disponibilidad del equipo en la ciudad. El diagnóstico definitivo se basa en el aspecto arteriográfico que se encuentra en la panangiografia cerebral2,3,9, tal y como se evidenció en nuestro caso, además la severidad se clasifica según el sistema de clasificación de Suzuki (tabla 2)3, donde nuestra paciente se encontraba dentro del estadio III de la enfermedad.
El tratamiento quirúrgico precoz es el de elección; existiendo distintas técnicas como la anastomosis de la arteria temporal superficial con la arteria cerebral media (ATS-ACM), encefalomiosinangiosis (EMS), encefaloduroarteriomiosinangiosis (EDAMS) y el trasplante del epiplón sobre la superficie cerebral9, al negarse el tutor legal de nuestra paciente a firmar el consentimiento informado, no fue posible realizar ningún tipo de intervención.
El pronóstico de los pacientes con esta patología no es totalmente predecible, sin embargo, el comienzo de la sintomatología antes de los 5 años de edad se ha relacionado con mal pronóstico; los cuadros hemorrágicos tienen un alto grado de mortalidad y recurrencia8,14, estando acorde con el desenlace final de nuestro caso.
En resumen, la EMM es más frecuente en mujeres de edad adulta (45-49 años), los síntomas dependen de las alteraciones del flujo sanguíneo cerebral. El diagnóstico definitivo se hace con panangiografía cerebral y el tratamiento quirúrgico es preferido que el médico, puesto que presenta mejor evolución y pronostico.
Conflicto de interés
Los autores manifiestan no tener conflictos de intereses en este estudio.
Referencias
1. Africano H, Moreno R. Reporte de dos casos de moyamoya en el Hospital Universitario. Los Comuneros de Bucaramanga (Santander). Acta neurológica Colombiana. Acta. Neurol. Colomb. 2015; 31(3):310- 317. [ Links ]
2. Scherle MC, et al. Síndrome de Moyamoya en un niño con Drepanocitosis. Hospital Hermanos Amejeiras Ciudad Hababa, Cuba. Rev Ecuatoriana de Neurología. 2011; 20: 1-3. [ Links ]
3. Arteaga J, Burgos M, Quintana L. Enfermedad Moyamoya. Presentación de un caso y revisión de la literatura. Rev. chil. neuropsicol. 2015; 41(64): 116-119. [ Links ]
4. Espinosa Ea, et al. Síndrome y enfermedad de Moyamoya. Act. Neurol. Colomb. 2011;27(7):3. [ Links ]
5. Resolución No. 2048/2011. Por la cual se actualiza el listado de enfermedades huérfanas. Boletín oficial N° 047. 9 de junio del 2015. [ Links ]
6. Ruiz HV., Hoyos Pulgarin JA. Enfermedad de Moyamoya: Reporte de caso y revisión de la literatura. Rev Med de Risaralda. 2012;18(2):172-178. [ Links ].
7. Ramírez-Quiñones JA, Barrientos-Imán DM, Calle-La RP, Ecos-Quispe RL, Novoa-Mosquera ME, Valencia-Chávez AM, et al. Enfermedad de Moyamoya: reporte de un caso. Rev Neuropsiquiatr. 2015;78(3):165-170. [ Links ]
8. Urrutia-Ruiz M, et al. Enfermedad de moyamoya. Bol Med Hosp Infant Mex. 2007; 64 (22):99-106. [ Links ]
9. Martínez Barros M, et al. Síndrome de moya-moya y falciformia. Reporte de un caso. Revista Facultad de Ciencias de la Salud . Duazary. 2012;9(2):176-180. [ Links ]
10. Lizarazo J, Niño F, Alvarado H, Castro N. Síndrome moyamoya y enfermedad de Graves en una mujer joven. Acta Med Colom. 2013;38(4): 262-267. [ Links ]
11. Vargas DJ, Garófalo GN, Barroso GE, et al. Enfermedad de moyamoya, macrocefalia y déficit intelectual en un adolescente. Rev Cubana Pediatr. 2013;85(1):112-119. [ Links ]
12. Moschini Javier. El signo de la hiedra: un patrón de realce leptomeníngeo difuso característico del síndrome de moyamoya. Rev Neurol Arg.2011;79(12):3. [ Links ]
13. Navarro-Ballester A, Ambit-Capdevila S, Pérez-Caballero FA., Marco-Domenech SF. Enfermedad moyamoya en un paciente con síndrome de Down. Anales de Radiología México. 2013;4(12):262-265. [ Links ]
14. Gutiérrez-Morales JL, Domínguez-Moreno R, Morales-Esponda M, Natalia Lorena, Rossiere-Echazarreta, Espinoza-Castilla A, Rodríguez-Guzmán LM. Enfermedad de moyamoya. Presentación de un caso. Gac Méd Méx. 2010;1(3):77-84. [ Links ]

