










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkActa Neurológica Colombiana
Print version ISSN 0120-8748
Acta Neurol Colomb. vol.32 no.4 Bogotá Oct./Dec. 2016
https://doi.org/10.22379/24224022115
https://doi.org/10.22379/24224022115
Caso clínico
Epilepsia y síndrome dismórfico asociado a cromosomopatía hereditaria ligada al cromosoma X, tipo micro-duplicación
Epilepsy and dysmorphic syndrome associated with hereditary chromosomopathy linked to X chromosome, micro-duplication type
Wolfang Rubio Rodríguez (1), Martín Torres Zambrano (2), Giancarlos Conde Cardona (3), Aldo Caraballo (4), Margarita García Meléndez (5), Luis Polo Verbel (6), José Andrés Gamero Tafur (7), Juan Torres Sandoval (8).
(1) Residente de Neurología clínica, cuarto año, Universidad del Sinú, Fundación Centro Colombiano de Epilepsia y Enfermedades Neurológicas, FIRE, Cartagena, Colombia
(2) Neurólogo, docente de posgrado Neurología, Fundación Centro Colombiano de Epilepsia y Enfermedades Neurológicas FIRE, Cartagena, Colombia
(3) Residente Neurología Clínica, tercer año, Universidad del Sinú, Fundación Centro Colombiano de Epilepsia y Enfermedades Neurológicas FIRE, Cartagena, Colombia
(4) Pediatra, Fundación Centro Colombiano de Epilepsia y Enfermedades Neurológicas FIRE, Cartagena, Colombia
(5) Neuropediatría, docente de posgrado Neurología, Hospital Infantil Napoleón Franco Pareja, Cartagena, Colombia
(6) Neurólogo, docente de posgrado Neurología, Fundación Centro Colombiano de Epilepsia y Enfermedades Neurológicas FIRE, Cartagena, Colombia
(7) Estudiante de Medicina, Corporación Universitaria Rafael Núñez, Cartagena, Colombia
(8) Médico general, Universidad del Rosario, Cartagena de indias, Colombia
Recibido: 20/06/16. Aceptado: 28/12/16.
Correspondencia: Wolfang Rubio Rodríguez, neuronavegador@hotmail.com
Resumen
Los hallazgos de síndromes dismórficos asociados a cromosomopatía ligada a X y epilepsia son de presentación infrecuente. Presentamos un caso de alteración genética en un paciente masculino, con microduplicación ligada al cromosoma X MECP2 y antecedente familiar de hermano con fenotipo similar, que comparten línea sanguínea materna, de diferentes padres. El síndrome dismórfico ligado a cromosoma X MECP2 (methyl-CpG-binding protein2), causan grave retraso mental, encefalopatía epiléptica e infecciones recurrentes del aparato respiratorio y consecuentemente pueden además tener una epilepsia resistente al manejo farmacológico.
Palabras clave: dismórfico, encefalopatía, epilepsia, genética, infección, intratable (DeCS).
Summary
The findings of dysmorphic syndromes associated with X-linked chromosomopathy and epilepsy are infrequent. It is a case of genetic alteration in a male patient, with X-linked microduplication MECP2 and familiar history of a sibling with similar phenotype, which compares the maternal blood line of different parents. X-linked dysmorphic syndrome MECP2 (methyl-CpG2 binding protein), causing severe mental retardation, epileptic encephalopathy and recurrent infections of the respiratory tract and consecutively also have epilepsy resistant to pharmacological management.
Key words: dysmorphic, encephalopathy, epilepsy, genetic, infection, intractable (MeSH).
Introducción
Las cromosomopatías ligadas al cromosoma X hacen parte de las etiologías de presentación en pacientes con déficit global neuromadurativo y epilepsia. El nivel de penetrancia y el número de microdeleciones implicadas generan espectros distintos de manifestación de la enfermedad. A continuación se describe un caso de síndrome dismórfico y epilepsia asociado a cromosomopatía hereditaria ligada a X, el cual presenta antecedente familiar de hermano por línea materna, que cursa con encefalopatía epiléptica y características fenotípicas idénticas.
Reporte de caso
Paciente masculino de 9 años, con diagnóstico de epilepsia generalizada de inicio a los 5 años, refractaria a manejo farmacológico, con parálisis cerebral espástica y retraso neuromadurativo global. Antecedentes personales de infecciones respiratorias recurrentes, requirió hospitalización y manejo. Al examen físico con medidas antropométricas: talla 138 cm - peso 24 kg - PC 53 M - IMC 13 kg/M2. Fenotipo (figura 1) con paladar ojival, mallampathy III, implantación auricular baja, pabellón auricular proyectado hacia adelante, implantación baja del cabello, hipoplasia malar bilateral, escapulas aladas, genu varo, pie plano. Se realizaron paraclínicos (tabla 1) y resonancia magnética nuclear 1.5 tesla (figura 2), en la que se reporta hipoplasia del cuerpo calloso, hipodesarrollo del vermis cerebeloso.
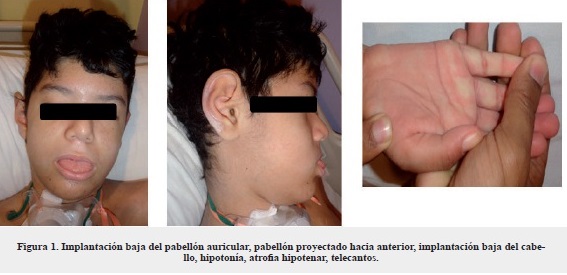

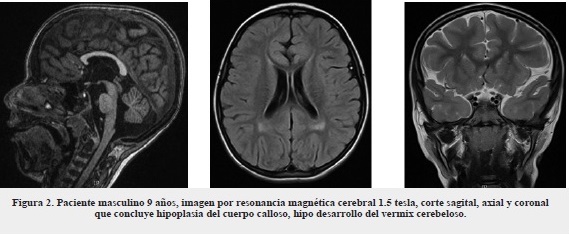
Antecedente familiar de hermano por línea materna con características fenotípicas similares a las descritas con diagnóstico de encefalopatía epiléptica refractaria al manejo farmacológico.
Teniendo en cuenta el cuadro clínico y antecedente, se consideran estudios para síndrome dismórfico en relación al cromosoma X. se realiza microarray de ADN bajo plataforma affimetrix cytoscan con micro duplicaciones en cr 3 y cromosoma X región q28. Se identifica duplicación en MECP2 lo que confirma la sospecha clínica para ambos pacientes.
Discusión
Los estudios estadísticos demuestran la importante relación que existe entre el déficit cognitivo y su importante relación con los trastornos genéticos relacionados con el cromosoma X. Para el año 2001 se registraron alrededor de 26 síndromes asociados a esta condición clínica con una incidencia en la población general de 1:150.000 individuos, a los cuales se les puede hacer identificación plena de la lesión genética. Además, la literatura reporta la identificación de aproximadamente 102 genes relacionados con este síntoma; algunos de ellos también relacionados con epilepsia; sin embargo, es claro entender que no todos los pacientes con epilepsia también cursan con retardo del neurodesarrollo y se debe ser muy puntual en establecer la alteración genética para poder estimar el nivel de perturbación de la estructura o función neuronal y determinar su evolución1.
La aparición de epilepsia resistente al manejo farmacológico en la primera infancia relacionada con alteraciones genéticas del cromosoma X es una patología infrecuente, sin embargo, hay descripciones de pacientes en los cuales se ha identificado retardo mental, síndrome dismórfico, fallas en el sistema inmune, síndromes atáxicos y variantes sindromáticas entre las que se describe síndrome de Angelman, síndrome de Rett, síndrome Lennox-Gastaut, síndrome de West y en más del 80 % de los casos, con alteraciones en el electroencefalograma (EEG) y diferentes tipos de crisis que podrían tener relación con este cuadro2,3.
Característicamente las crisis que se presentan están dadas por convulsiones tónico-clónicas generalizadas que frecuentemente se desarrollan dentro de los primeros dos años de vida. No es inhabitual que estos casos posteriormente sean seguidos por cuadros de ausencia atípica, crisis atónica y crisis parciales complejas. Con una respuesta pobre a la terapia anticonvulsiva y un trazado de electroencefalografía que muestra actividad rítmica lenta de gran amplitud, típica del síndrome de Angelman en algunos pacientes, pero no en otros. Las neuroimágenes muestran atrofia o hipoplasia de estructuras cerebrales con progresión en el tiempo4.
A pesar de la identificación de grupos familiares con estas cromosomopatías, las alteraciones por microdeleciones del cromosoma X son una patología mucho más infrecuente y típicamente relacionada con epilepsia generalizada que finaliza en cuadros de encefalopatía epiléptica y alteraciones del sistema inmunológico, que facilitan la aparición de procesos infecciosos, particularmente de la vía respiratoria y de tejidos blandos; el estudio de las alteraciones por microarreglos ha demostrado una gran constelación de causas como substrato de epilepsia, entre los cuales se encuentra los trastornos embriológicos en los procesos de proliferación, organización, diferenciación y migración neuronal puestos en evidencia en los focos heterotópico en substancia blanca y en nidos de celularidad con una inadecuada organización o heretotipia celular, algunos de ellos con cambios que se pueden traducir en variaciones tan drásticas como lisencefalia, polimicrogiria o microcefalia5.
Dentro de los cambios proclives a la conformación de las alteraciones de la microarquitectura celular, también se describen alteraciones a nivel de las cadenas de actina que conforman el citoesqueleto que posteriormente constituirán el andamiaje y las zonas de anclaje para la adecuada diferenciación de las cortezas que conforman cada uno de las áreas cerebrales. Los cambios a este nivel también se traducen en la aparición de alteraciones morfológicas y en consecuencia focos con celularidad epileptógena, que pueden desarrollar descargas que hagan parte de los hallazgos en el contexto de la epilepsia farmacológicamente intratable de estos pacientes. Otro mecanismo por el cual se presenta este tipo de manifestación clínica es la alteración del medio en el cual se encuentran las células y la variación en los gradientes iónicos que las circundan, esto trae como consecuencia una expresión inadecuada de canales iónicos o serias deficiencias en la respuesta al gradiente iónico por alteraciones en las concentraciones habituales a las que están sometidas las células heterotópicas6.
Teniendo en cuenta que la gran mayoría de los casos se relacionan con fenómenos infecciosos recurrentes, esta característica no es ajena al caso en mención, se debe explicar que la susceptibilidad obedece a una falla en los niveles de anticuerpos y más exactamente para aquellos que deberían estar presentes en la vía área para la protección contra patógenos frecuentes como el caso de S. Pneumoniae y otros agentes relacionados con procesos infecciosos de tejidos blandos como Staphyloccocus Aureus6.
Por otra parte el gen MECP2 está directamente relacionado con la inadecuada expresión de las células L1 de adhesión que conforman la molécula L1CAM, gen que se ha relacionado directamente con las alteraciones del fenotipo y retardo mental, la transmisión de esta anomalía está directamente relacionada con el cromosoma X y la razón por la cual la aparición de el retardo en neuromaduración es menor en mujeres obedece a la inactivación selectiva de la información génica por el X sano heredado, situación que no favorece a los hombres; sin embargo, algunos reportes de caso han permitido identificar casos aislados de presentación en mujeres que cumplen tanto con los hallazgos fenotípicos como las características clínicas de presentación de la enfermedad6,7.
El fenotipo del compromiso por alteraciones ligadas al cromosoma X es característicamente de predominio facial frente ancha y amplia, cabello rizado, telecantos y epicantos cejas finas muy delgadas, nariz pequeña, mejillas llenas, boca habitualmente abierta con labio inferior prominente, diastemas e hipertrofia gingival, pabellón auricular, implantación baja y discretamente puntiagudo, implantación baja del cuero cabelludo y cuello corto7,8.
En la búsqueda por establecer patrones de diferenciación se realizaron estudios poblacionales de familias en Noruega, Suiza, Ucrania, Reino Unido y EE. UU., encontrando la mutación MECP2, logrando identificar diferentes clases de compromiso y aspectos de presentación relacionados incluso con presentaciones en mosaico, en las cuales había compromiso en porcentajes parciales de la población estudiada, estos hallazgos nos llevan a concluir que existe un gran polimorfismo de presentación del cuadro clínico y como la variación por microdeleciones puede llegar a afectar a diferentes niveles, la expresión de dominios celulares transmembrana y poner de manifiesto una expresión distinta de la enfermedad3, hecho tal que reforzaría la teoría sobre la expresión genómica de patrones de mosaicismo en los cuales la penetrancia y modifica la expresión en el paciente, como se evidenció en el estudio de poblaciones mexicanas, en donde se hizo seguimiento para casos de síndrome de Lubs9,10.
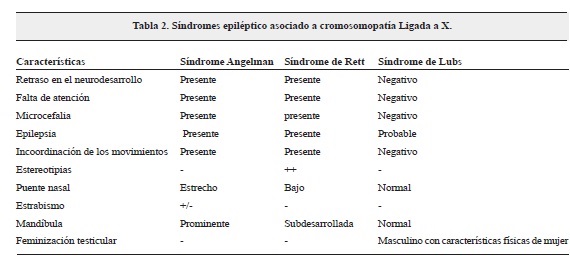
Al realizar una búsqueda en las descripciones de caso de cromosomopatías asociadas con el cromosoma X (tabla 2), las características descritas para algunas de ellas son claras; microcefalia del síndrome de Rett, la cual no está presente en nuestros casos, o la facies, característica de felicidad y el patrón conductual del Angelman en las cuales definitivamente no se hacen evidentes para los pacientes descritos. Finalmente nuestros casos, tampoco comparten ninguna de las características con el Lubs, en el que el paciente tiene rasgos típicos asociados a la baja respuesta hormonal, por otra parte las duplicaciones del MECP2 con los rasgos característicos de hipotonía, retardo severo del neurodesarrollo y espasticidad progresiva, pero a diferencia de nuestros casos, la descripción infrecuente de encefalopatía epiléptica, situación que difiere de nuestra casuística al igual que los rasgos fenotípicos, tampoco corresponden con total similitud a los rasgos de los pacientes en mención.
Conclusión
Se describen los casos de dos hermanos con fenotipo idéntico, retardo mental y encefalopatía epiléptica, los cuales se presentan en el contexto de una cromosomopatía ligada al cromosoma X, tipo microduplicación; al hacer la búsqueda en la literatura se han encontrado diferentes patrones de presentación de la enfermedad, sin embargo, las características físicas descritas para los síndromes en mención no corresponden a otros ya mencionados en el estudio Angelman, Rett y Lubs, lo que deja abierta la posibilidad de que esta descripción corresponda a otro síndrome.
Conflicto de interés
Los autores manifiestan no tener conflictos de intereses en este estudio.
Referencias
1. Prescott TE, Rødningen OK, Bjørnstad A, Stray-Pedersen A. Two brothers with a microduplication including the MECP2 gene: rapid head growth in infancy and resolution of susceptibility to infection: Clin Dysmorphol. 2009 Apr;18(2):78-82. [ Links ]
2. Gilfillan GD, Selmer KK, Roxrud I, Smith R, Kyllerman M, Eiklid K, et al. SLC9A6 Mutations Cause X-Linked Mental Retardation, Microcephaly, Epilepsy, and Ataxia, a Phenotype Mimicking Angelman Syndrome. Am J Hum Genet. 2008 Apr;82(4):1003-10. [ Links ]
3. Moog U, Roozendaal KV, Smeets E, Tserpelis D, Devriendt K, Buggenhout GV, et al. MECP2 mutations are an infrequent cause of mental retardation associated with neurological problems in male patients. Brain Dev. 2006 Jun;28(5):305-10. [ Links ]
4. Stevenson RE, Holden KR, Rogers RC, Schwartz CE. Seizures and X-linked intellectual disability. Eur J Med Genet. 2012 May;55(5):307-12. [ Links ]
5. Sherr EH. The ARX story (epilepsy, mental retardation, autism, and cerebral malformations): one gene leads to many phenotypes. Curr Opin Pediatr. 2003 Dec;15(6):567-71. [ Links ]
6. Hirose S, Mitsudome A. X-Linked mental retardation and epilepsy: pathogenetic significance of ARX mutations. Brain Dev. 2003 Apr;25(3):161-5. [ Links ]
7. Bijlsma EK, Collins A, Papa FT, Tejada MI, Wheeler P, Peeters EAJ, et al. Xq28 duplications including MECP2 in five females: Expanding the phenotype to severe mental retardation. Eur J Med Genet. 2012 Jun;55(6-7):404-13. [ Links ]
8. Giannandrea M, Bianchi V, Mignogna ML, Sirri A, Carrabino S, D'Elia E, et al. Mutations in the Small GTPase Gene RAB39B Are Responsible for X-linked Mental Retardation Associated with Autism, Epilepsy, and Macrocephaly. Am J Hum Genet. 2010 Feb;86(2):185-95. [ Links ]
9. Neira VA, Romero-Espinoza P, Rojas-Martínez A, Ortiz-López R, Córdova-Fletes C, Plaja A, et al. De novo MECP2 disomy in a Mexican male carrying a supernumerary marker chromosome and no typical Lubs syndrome features. Gene. 2013 Jul;524(2):381-5. [ Links ]
10. Mencarelli MA, Katzaki E, Papa FT, Sampieri K, Caselli R, Uliana V, et al. Private inherited microdeletion/microduplications: Implications in clinical practice. Eur J Med Genet. 2008 Sep;51(5):409-16. [ Links ]

