











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkDe acuerdo con la nueva clasificación de la Movement Disorders Society, explicada en el capítulo 1 de este consenso, la distonía se clasifica según su etiología en hereditaria, adquirida e idiopática. La distonía primaria se definía como aquella única manifestación fenotípica, sin embargo, suele acompañarse de otros trastornos del movimiento, por lo que se crearon dos términos que se ajustan más a los nuevos avances en la genética y clasificación 1-4.
Distonía aislada: donde la única manifestación es la distonía, aunque se acepta que se acompañe de temblor distónico.
Distonía combinada: donde además de la distonía se encuentran otras alteraciones del movimiento como mioclonus y parkinsonismo. Este término reemplaza al antiguo distonía-plus.
La distonía hereditaria, tema que trataremos en este capítulo, tiene una gran variedad de presentaciones clínicas, así como de genes asociados (tabla 1). A medida que avanzan los estudios genéticos, muchas de las distonías idiopáticas ya sean familiares o esporádicas, serán incluidas como genéticas, conforme ha ocurrido en los últimos años.
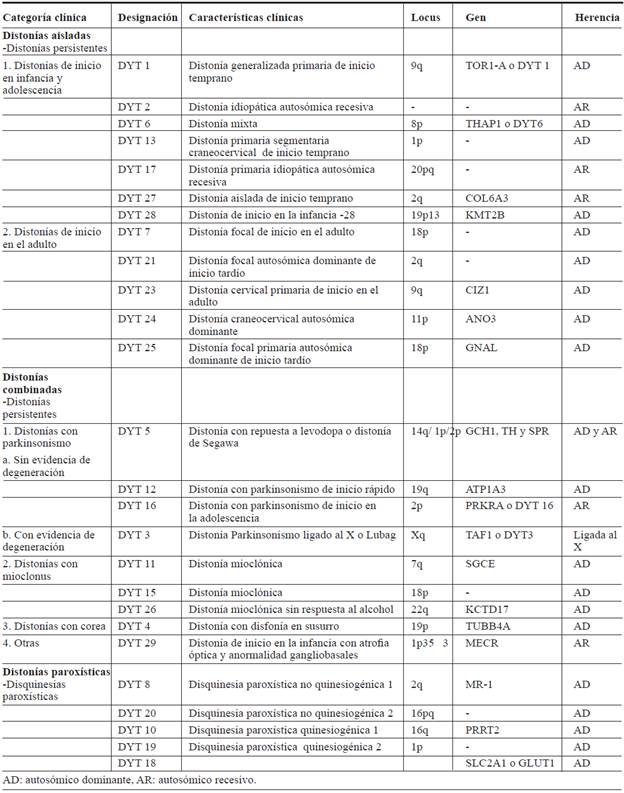
La nomenclatura de las distonías genéticas es secuencial de acuerdo con la fecha en que se describieron, por lo que se organizará según sus características clínicas y edad de inicio, los parámetros de la nueva clasificación del 2013 3-5.
DISTONÍAS AISLADAS DE INICIO EN LA INFANCIA-ADOLESCENCIA 3-7.
DYT 1. Distonía primaria generalizada de inicio temprano (distonía muscular deformante o enfermedad de Openheimer), (DYT-TOR1A)
En 1989 se logró ubicar su locus genético en el cromosoma 9q34 y se confirmó que está relacionado con la mayoría de distonías de este tipo en las familias estudiadas. Luego, en 1997 se localizó la mutación causal, siendo ésta una deleción GAG en el gen que se codifica para una proteína llamada torsina A, la cual está compuesta de 5 exones, la mutación se localiza en la posición 946 del exón 5. Esta mutación causa una pérdida de un residuo de ácido glutámico cerca del extremo carboxilo terminal, esta proteína es una ATPasa que está relacionada con la regulación sináptica y la respuesta al estrés celular. Esta proteína se distribuye difusamente en el cerebro, con intensa expresión en las neuronas dopaminérgicas de la sustancia nigra compacta, núcleo dentado del cerebelo, neuronas de Purkinje, locus ceruleus, base del puente, numerosos núcleos talámicos, núcleos pendunculopontinos, núcleos oculomotores, formación hipocampal y corteza frontal, lo que sugiere un predominio de disfunción dopaminérgica.
Esta distonía se caracteriza por tener una herencia autosómico dominante, con penetrancia (pacientes con presencia de la mutación sin enfermedad) del 30 al 40 %, lo que sugiere que pueden existir otros factores ambientales y/o genéticos que se relacionan con la expresión fenotípica.
Clínicamente se ha observado una importante variabilidad en los afectados, pero hay dos características clínicas consistentes:
Inicio de síntomas antes de los 20 años
Compromiso inicial de extremidades, principalmente las piernas
Actualmente se recomienda estudiar la presencia del DYT 1 en pacientes menores de 30 años, con distonía que inicie por una de las extremidades.
DYT 2
Es una distonía con patrón de herencia autosómico recesivo, con pocos casos descritos, donde no se ha encontrado un locus genético aún. Sus características clínicas son similares a la DYT 1 y se ha encontrado en familias consanguíneas.
DYT 6. (DYT-THAP1) 8
Es una distonía autosómica dominante, con una penetrancia estimada en el 60 %. El gen alterado THAP1 (Thanatos-associated protein domain containing apoptosis associated protein 1) formado por 3 exones, codifica una proteína nuclear pro-apoptosis, cuya función es potenciar la apoptosis inducida por el factor de necrosis tumoral (TNF) alfa. Estas proteínas también están relacionadas con la regulación de la proliferación celular y se cree que generan la distonía a través de un mecanismo de pérdida de función.
Las principales formas de presentación son una distonía generalizada de inicio en la adolescencia, seguida de una forma segmentaria. Sin embargo, otros estudios la han relacionado con distonías focales, en especial la distonía cervical de inicio tardío. Otra característica que la diferencia de la DYT 1, es que cuando comienza en las extremidades tiende a predominar en los miembros superiores.
DYT 13
Tiene una herencia autosómica dominante, con una penetrancia estimada en el 58 %. Se ha descrito únicamente en una familia italiana, el gen causante no ha sido descrito aún pero se ha mapeado su locus en el brazo corto del cromosoma 1. Su edad de inicio varía entre los 5 y 40 años, con promedio de 15,6 +/- 12,5 años. En la mayoría de los casos hay compromiso cráneo-cervical y de miembros superiores, con lenta progresión y sólo se generaliza en el 18.2 % de los casos. Su fenotipo es similar a la DYT 6 pero con menor compromiso laríngeo y de miembros inferiores.
DYT 17 9
Se ha descrito en una familia libanesa, con 3 hermanas afectadas, que muestran un patrón de herencia autosómico recesivo. Se presenta como una distonía cervical entre los 14 y 19 años, que en 2 casos evolucionó a segmentaria luego de 2 a 3 años del inicio y a generalizada en el otro, luego de 9 años. Su gen se ha mapeado en el cromosoma 20 (20p11.22-q13.12).
DYT 27. (DYT-COL6A3) 10
Descrita en familias alemanas con patrón de herencia autosómico recesivo, asociado a una mutación del gen COL6A3, que codifica la cadena a-3 del colágeno tipo VI, no es claro el mecanismo por el cual se genera esta patología.
Inicia en las 2 primeras décadas de la vida, como una distonía focal o segmentaria de la región craneocervical o de miembros superiores. Se puede observar temblor distónico de acción o postural, calambre del escribano, distonía oromandibular o distonía laríngea.
DYT 28. (DYT-KMT2B) 11
Se caracteriza por ser una distonía autosómico dominante, inicia en la primera década de la vida (3 a 11 años), con compromiso inicial de miembros inferiores y posterior generalización con compromiso de miembros superiores, cuello y región orofacial, su severidad es variable y algunos pacientes terminan en silla de ruedas. Algunos pacientes presentan cara elongada y nariz bulbosa, cerca de la mitad de los afectados tienen retardo de desarrollo psicomotor, con compromiso cognitivo leve, también pueden haber alteraciones del movimiento ocular. Se han descrito diferentes mutaciones del gen KMT2B (19p23) que codifica una metiltransferasa específica para lisina, que metila la histona 3 y está relacionada con procesos de activación génica. En estudios de cultivos celulares se ha encontrado reducción en la expresión de THAP1 y TOR1 (ver DYT 6 y 1), lo que sugiere que este gen afecta la expresión de otros genes asociados a distonía. Las neuroimágenes muestran lesiones del globo pálido en la mayoría de pacientes.
DISTONÍAS AISLADAS DE INICIO EN EL ADULTO 3-8
DYT 7
Originalmente descrita en una extensa familia alemana, su gen se ha mapeado en el brazo corto del cromosoma 18, tiene una herencia autosómica dominante con penetrancia incompleta. Su presentación clínica es de distonía cervical, que puede tener compromiso leve facial, de miembros inferiores y disfonía espasmódica. Estudios recientes de familias con fenotipo similar y de la misma familia inicial, han fallado en encontrar alteraciones del cromosoma 18, por lo que puede haber heterogeneidad genética o aún no se encontrado el locus definitivo.
DYT 21
Descrita en una familia sueca, con herencia autosómica dominante, con penetrancia del 75 al 90 %. Sus síntomas frecuentes son: blefaroespasmo, distonía cervical, distonía de miembros superiores; algunos casos pueden tener dis-fonía espasmódica. En los 16 pacientes descritos, 6 son generalizados, 7 multifocales, 2 segmentarios y 1 focal. Su locus se ha mapeado al cromosoma 2 (2q14.3-21.3).
DYT 23. (DYT-CIZ1)
Es la primera familia con distonía cervical descrita, con un patrón de herencia autosómico dominante, en el seguimiento a largo plazo ningún caso ha evolucionado a distonía generalizada. Se ha encontrado una mutación de ensamblaje del potenciador en el exón 7 del gen CIZ1, localizado en el cromosoma 9, que codifica una proteína (Cip1-interacting zinc finger protein 1) que participa en la replicación del ADN y regulación del ciclo celular, y que tiene patrones de expresión similares al de la DYT 1 y 6.
DYT 24. (DTY-ANO3)
Es una distonía autosómica dominante, cuya edad de inicio puede estar entra la infancia y la quinta década, su inicio generalmente es en la región cervical, posteriormente hay distonía laríngea y compromiso de miembros superiores; nunca hay compromiso de miembros inferiores. La característica más consistente es la presencia de temblor distónico y en algunos pacientes puede observarse mioclonías. Se han encontrado en la familia descrita al menos 6 mutaciones del gen ANO3, que codifica para un canal de cloro activado por calcio y que tiene una alta expresión en el estriado.
DYT 25. (DYT-GNAL)
Es de patrón autosómico dominante, se han encontrado 8 familias con mutación del gen GNAL (proteína G subunidad alfa L) como su causa. Clínicamente se presenta inicialmente como una distonía cervical, con tendencia a pasar a ser craneocervical, con disfonía espasmódica con o sin generalización.
DISTONÍAS COMBINADAS CON PARKINSONISMO, SIN EVIDENCIA DE DEGENERACIÓN 3-9
Distonía con respuesta a levodopa (DRD) DYT 5a (DYT/PARK-GCH1), DYT-5b (DYT/PARK-TH)
Esta distonía tiene un patrón de herencia autosómico dominante en la mayoría de los afectados, es secundaria a mutaciones que se han descrito en el gen GCH1, y que codifica para la enzima GTP ciclohidrolasa. Hay una forma clínicamente similar pero con herencia autosómico recesiva, con mutaciones en el gen de la tirosina hidroxilasa (TH), por este motivo se propone dividir esta distonía en 2 grupos DRD 1 (DRD-a o DTY5-a) y DRD 2 (DRD-b o DTY5-b) de acuerdo al gen causante.
La penetrancia de esta patología es de aproximadamente 30 %, pero si se toman en cuenta las formas atípicas puede variar entre el 38 y 100 %, es mayor en mujeres (87-100 %), que en los hombres (38-55 %). Todos los afectados con alteraciones del gen GCH1 son heterocigotos para el gen afectado, los homocigotos tienen una presentación clínica diferente.
El fenotipo clínica de la DYT 5 característico es la presencia de distonía que se asocia a parkinsonismo desde el inicio o durante el curso de la enfermedad, con empeoramiento del síntoma durante el curso del día en el 77 % de los casos y presenta una dramática mejoría con la administración de levodopa, usualmente a dosis bajas, la respuesta se observa en pocos días. Cuando aparecen corea o disquinesias al aumentar la dosis de levodopa buscando una mejor respuesta, es un indicador de refractariedad al tratamiento. Rara vez se observan disquinesias o fluctuaciones motoras a largo plazo.
En los casos secundarios a mutación del gen de la TH, los casos de homocigotos son más severos, tienen menor respuesta a levodopa y mayor riesgo de desarrollar disquinesias.
Recientemente se ha incluido dentro de la DYT 5 una mutación del gen de la sepiaptenina reductasa (SPR) como causa de esta distonía, con patrón de herencia autosómico recesivo.
Todas las enzimas afectadas participan en la cadena de producción de la dopamina, por lo que sus características clínicas son similares. La distonía DYT 14, que en clasificaciones previas esta como una entidad diferente, es causada por una mutación del gen GCH1, por lo que hoy en día hace parte de la DYT 5.
DYT 12. Distonía - parkinsonismo de inicio rápido (DYT/PARK-ATP1A3)
En una patología poco común con patrón de herencia autosómico dominante, con penetrancia reducida. El gen comprometido es ATP1A3, que codifica para sodio - potasio ATPasa a-3, la cual es una unidad catalítica de la bomba de sodio, se han descrito varias mutaciones de este gen, pero su mecanismo fisiopatológico no es claro.
Su inicio es en adolescentes o en adultos jóvenes, como una distonía con parkinsonismo, asociado de inicio súbito, que se desarrolla en minutos a días y está desencadenada por estrés psicológico. En pocas semanas deja de progresar, generalmente no hay mejoría o es muy leve. En algunos pacientes se puede observar un nuevo episodio de deterioro, luego de 1 a 9 años del inicial. Compromete extremidades y cara con disartria y disfagia, con un claro predominio rostrocaudal, donde los síntomas bulbares son más severos e inician antes que los de las extremidades superiores y a su vez estos son más severos que los de las extremidades superiores. En raros casos un parkinsonismo aislado puede iniciar antes de la distonía. Se han descrito problemas psiquiátricos en las familias con esta entidad, como depresión, trastornos de personalidad, ansiedad, pánico y fobia social.
Se ha encontrado que algunas familias con hemiplejia alternante de la infancia con herencia dominante, son causadas por mutaciones del mismo gen que genera esta distonía, las cuales son diferentes a las de la DTY 12.
DYT 16. (DYT-PRKRA)
Fue descrita en familias brasileñas, con un patrón de herencia autosómico recesivo, causadas por una mutación del gen PRKRA que codifica la proteína activadora de la proteína quinasa inducida por interferón EIF2AK2.
Los síntomas inician entre los 2 y 18 años, con compromiso focal de extremidades que ocasionan alteraciones de la marcha y escritura, con posterior generalización. También se observa sonrisa sardónica distónica, disartria, disfagia y cambios psiquiátricos. En la mayoría de los afectados se encuentran signos piramidales. Una característica de esta distonía es la extensión a cara, cuello o laringe, lo cual es raro en otras distonías como la DYT 1. Por lo menos la mitad de los pacientes presenta parkinsonismo, con bradiquinesia, temblor postural y congelamiento de la marcha, hay mala respuesta a levodopa o anticolinérgicos.
DISTONÍAS COMBINADAS CON PARKINSONISMO, CON EVIDENCIA DE DEGENERACIÓN 3-9
DYT 3. Distonía-parkinsonismo ligado al x o Lubag (DYT/PARK-TAF1)
Esta distonía se observa en hombres filipinos de la isla Panay, con mutación del gen TAF1 (cambios de secuencia), que funciona como un regulador del ciclo celular. Inicia como una distonía focal en extremidades (47 %), en particular en miembros inferiores (33 %), o como una distonía craneal con blefaroespasmo, distonía oromandibular (27 %). Con el tiempo hay extensión a otras áreas corporales pasando a ser segmentaria, multifocal o generalizada. Después de 2 años de enfermedad el 84,7% de los pacientes han desarrollado distonía generalizada. Después de unos 5 a 7 años de evolución, la distonía tiende a revertir, después de 10 años sólo el 8 % de los pacientes tiene distonía, la cual es reemplazada por bradiquinesia progresiva y rigidez.
En estudios patológicos se ha encontrado pérdida de neuronas gabaérgicas del estriado, primero con compromiso de los estriosoma y posteriormente de la matriz, lo que puede explicar el inicio con distonía y el posterior paso a parkinsonismo.
DISTONÍAS COMBINADAS CON MIOCLONÍAS Y COREA 5-9.
DYT 11. Distonía mioclónica (DYT-SGCE)
Es una entidad con patrón de herencia autosómico dominante, con penetrancia variable, secundario a mutación del gen SGCE, que codifica para la epsilon - sarcoglicano, que es una proteína transmembrana que forma parte del complejo glicoproteico de la distrofina presente en el músculo esquelético y cardiaco, en el sistema nervioso, en las neuronas monoaminérgicas, las células de Purkinje, la corteza cerebral y el hipocampo.
El cuadro clínico típico inicia entre la primera y segunda décadas de la vida, con la presencia de mioclonías, que pueden ser aisladas o asociarse a una distonía leve a moderada, que se localiza principalmente en la mitad superior del cuerpo, las formas más comunes son la distonía cervical y el calambre del escribano. Se pueden ver casos de distonía sin mioclonías al inicio pero con el tiempo estas aparecen. Típicamente hay mejoría con alcohol etílico. Se asocia a cambios psiquiátricos como depresión, trastorno obsesivo compulsivo, trastornos de personalidad, déficit de atención con hiperactividad y abuso de drogas.
DYT 15
Su clínica es prácticamente idéntica a la DYT 11, pero no se encontró mutación en el gen SGCE, aún no se conoce su causa, pero se ha mapeado su gen al brazo corto del cromosoma 18.
DYT 4. (DYT-TUBB4A)
Fue descrita en una familia australiana, con patrón de herencia autosómico dominante con penetrancia completa, secundaria a una mutación del gen de la tubulina B (proteína del citoesqueleto). Su edad de inicio está entre los 13 y 37 años, pero suele ser antes de los 20. Hay una importante variación en su expresión que puede ir de una distonía en susurro, hasta fenotipos similares a una corea de Hunting-ton. Es una enfermedad progresiva, que se asocia a cambios neuropsiquiátricos y a otros desórdenes del movimiento como corea y ataxia.
Otra mutación del gen de la tubulina B produce leucoencefalopatía, hipomielinización con atrofia de los ganglios basales y el cerebelo (H-ABS), en la que hay distonía, retardo del desarrollo psicomotor, espasticidad, ataxia, disartria, estatura corta y microcefalia, se ha encontrado superposición fenotípica en ambas entidades, siendo más severa y de inicio más temprano la H-ABS.
DYT 26. Distonía mioclónica sin respuesta al alcohol (DYT-KCTD17) 12
Esta es una distonía asociada a mioclonías, con herencia autosómica dominante, generada por mutaciones en el gen KCTD17 en el cromosoma 22. Su clínica es similar a la DYT 11, tiende a ser progresiva con mayor compromiso de la mitad superior del cuerpo, pero en la DYT 26 no se observa respuesta al alcohol.
DYT 29. (DYT-MECR) 13
Esta entidad tiene un patrón autosómico recesivo y sus síntomas empiezan entre los 15 meses y 7 años de vida, con movimientos anormales, principalmente distonía, en algunos casos puede haber retardo leve del desarrollo psicomotor, como características variables se observan: distonía facial, espasticidad de miembros inferiores, hiperreflexia, corea, mioclonus, disquinesias, disartria y disfagia. La gran mayoría progresa con importante dificultad para la marcha. La atrofia óptica con disminución de agudeza visual inicia inmediatamente o pocos años después de la distonía, pueden observase alteraciones oculomotoras y nistagmus. Las imágenes muestran hiperintensidad en T2 de los ganglios basales y se puede observar un pico de lactato en la espectroscopia. A pesar de ser una enfermedad progresiva, parece no tener deterioro cognitivo asociado. Esta enfermedad se considera un error innato del metabolismo, ya que la enzima codificada por este gen participa en la síntesis del ácido lipoico y es requerida para la competencia energética de la mitocondria, por esto compromete órganos con altas demandas energéticas y alta susceptibilidad al estrés oxidativo, como los ganglios basales y el nervio óptico.

