











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink"Imagina los síntomas del autismo, la parálisis cerebral, la enfermedad de Parkinson, la epilepsia y el trastorno de ansiedad...
todo en una misma niñita"
Rett Syndrome Research Trust
DESCRIPCIÓN METODOLÓGICA DE LA BÚSQUEDA DE CASOS Y REVISIÓN DEL TEMA
Entre el primer semestre de 2014 y septiembre de 2016, se buscaron investigaciones, reportes y presentaciones de caso del síndrome de Rett, sin filtro cronológico, con las palabras clave "Rett", "Rett Syndrome", "Rett Syndrome review", "Rett syndrome therapy", "Rett cases", "Colombia", "America", "MECP2" y 'Rettprevalence", en las bases de datos en línea: Biblioteca Virtual en Salud (BVS), Medic Latina, Directory of Open Access Journals (Doaj), Embase, Biomed Central, EBSCOhost, FreeFullPDF, UpToDate, Journal Storage (Jstor), Medline, Imbiomed, Pubmed, PMC, MedGen, OMIM, Human Gene Mutation Database (HGMD), RettBASE, Eeiden Open Variation Database LOVD, OVID-Journals, Scientific Electronic Library Online (SciELO) y Science Direct; en las revistas electrónicas o medios físicos: Nature, Nature Neuroscience, Nature Reviews Neuroscience, Journal of Basic and Applied Genetics, Revista de Ciencias de la Salud, Revista Biomédica, Revista Univérsitas Médica, Revista Médicas UIS, Revista Salud UIS, Revista MedUNAB, Revista Colombia Médica, Acta Médica Colombiana, Acta Neurológica Colombiana y Revista Pediatría, entre otras; en la página oficial del Rett Syndrome Research Trust (reverserett.org/research) y en los motores de búsqueda generales: Yahoo® y Google®.
Los textos encontrados fueron clasificados según la relevancia sentida por los autores en los títulos y resúmenes, con prioridad para los artículos originales en los idiomas inglés y español posterior al año 2006. Además, se revisaron todos los manuscritos con información sobre el síndrome de Rett publicados en países latinoamericanos y los de tipo reporte de caso o de actualización epidemiológica sin filtros de fecha ni idioma. De manera concomitante, se buscó información adicional y la afiliación de familias colombianas en los portales oficiales en inglés y español de la International Rett Syndrome Foundation (IRSF) - (http://www.Rettsyndrome.org y http://www.rettsyndrome.org.es).
Los casos presentados en este artículo fueron captados y diagnosticados en la consulta del servicio de genética clínica realizada por los autores en la ciudad de Bucaramanga entre los años 2012 y 2016, con cobertura por seguridad social de la región de Santander, que incluye a los departamentos de Santander y Norte de Santander en Colombia. Se firmó consentimiento informado en cada caso. A todos los casos se les garantizó la confidencialidad de la información mediante la custodia de sus historias clínicas, la identificación mediante un código alfanumérico de conocimiento exclusivo de los autores, la garantía de no acceso por terceros y la eliminación de aquellos datos que pudieran generar una distinción o facilitar la búsqueda e identificación de las pacientes, según la normatividad ética y legal nacional e internacional vigente para estudios con humanos. Se tuvo en cuenta lo estipulado en el Título III, Categorías de datos sensibles, artículo 5° y 6° numeral e.) de la Ley estatutaria 1581 del 2012 que reglamentó el artículo 15 de la Constitución Política, en la que se establece que los datos relativos a la salud se consideran sensibles y se prohíbe su tratamiento excepto cuando el tratamiento de los datos tenga una finalidad histórica, estadística o científica.
INTRODUCCIÓN
El síndrome de Rett (RTT) [OMIM 312750] 1, es una condición neurológica severa de origen genético, que afecta de manera casi exclusiva al sexo femenino, con una pérdida progresiva de las ganancias psicomotoras en un historial de desarrollo normal 2-5. Aunque existen formas atípicas de la enfermedad, los hallazgos clínicos característicos son: pérdida de habilidades comunicativas y de las metas del desarrollo motor, ataxia o distaxia, tendencia autista, estereotipias, microcefalia y convulsiones 6-10. Se incluye en la cuarta versión revisada del Manual Diagnóstico y Estadístico de los Trastornos Mentales o DSM-IV-TR por sus siglas en inglés, dentro de los Trastornos Generalizados del Desarrollo (F84.2 Trastorno de Rett [299.80]) 11, por la afectación de las habilidades motora, cognitiva y de comunicación 12,13. Por su parte, en el DSM-5 publicado en 2013, referente a los Trastornos del Espectro Autista 14, se reconocen por presentar un déficit persistente de habilidades sociales y comunicativas, intereses limitados y movimientos estereotipados 15-18.
Pese a estar dentro del grupo de las "enfermedades raras", el RTT es la segunda causa genética de trastorno cognitivo profundo en mujeres, luego del síndrome de Down, afectando a todas las razas y reportándose en más de 40 países 2,8,12,19-21. Aun así, no se ha desarrollado un manejo específico ni se ha impactado en el pronóstico de estas pacientes, estimándose una esperanza de vida de 35 años 20, aunque se han descrito algunas pacientes que superan la sexta década de vida 3.
No obstante, se puede considerar como desconocido el estado actual de la enfermedad en Latinoamérica y particularmente en Colombia, dada la ausencia de reportes locales y estudios sistemáticos que involucren a dichas poblaciones. Se hace una revisión general del tema y se presenta la primera serie de casos documentada hasta la fecha en Colombia y la mayor en el continente, constando de siete niñas con RTT clásico y tres casos con RTT atípico, diagnosticadas por criterios clínicos y confirmados por prueba molecular.
CONTEXTO HISTÓRICO
Las primeras descripciones del RTT datan de la década del sesenta, cuando un neurólogo pediatra, Andreas Rett, evidenció en Alemania pacientes que desarrollaron regresión mental a edad temprana con movimientos estereotipados de las manos, oligofrenia grave e hiperamonemia 12,22-24. Aunque el médico desconocía que se tratara de una entidad particular, en 1977 publicó una serie de 21 niñas con los síntomas característicos bajo el título de "Cerebral Atrophy and Hyperammonaemia in childhood"12,13,22; posteriormente se reportaron casos similares en Japón, Inglaterra, Suecia, Portugal y Francia 25. En 1981, Bengt Hagberg describió una serie de 35 casos en el Manchester Meeting on Child Neurology, donde se considera el epónimo de RTT para esta entidad, propuesta aceptada en 1983 y que le caracterizaba como una enfermedad progresiva de autismo, demencia, pérdida del uso de las manos y aparición de movimientos estereotipados 1,12,22. Muy pronto aparecieron organizaciones como la IRSF y el Rett Syndrome Research Trust (RSRT), que reunieron grupos de apoyo para las familias de las pacientes en todo el mundo, recopilaron los avances científicos y estimularon investigaciones para desarrollar tratamientos.
En 1999 se relacionó con la enfermedad al gen codificante Methyl-CpG-binding protein 2 o MECP2, localizado en el cromosoma X, con el posterior descubrimiento de otros genes y mutaciones posiblemente relacionados con RTT1,6,20,26-28. Para el 2001, se reunió en Baden-Baden, Alemania, The Rett Syndrome Diagnostic Criteria Work Group, que concertó y publicó una lista de criterios diagnósticos clasificándolos como necesarios, de soporte y de exclusión 20. Sin embargo, en la práctica estos criterios resultaron ser muy extensos, poco específicos e incluso confusos para el estudio y el tratamiento de las pacientes, por lo que en 2010, el RettSearch Consortium revisó y publicó una nueva nomenclatura y criterios diagnósticos para el RTT que todavía permanecen vigentes y que se aplicaron para el diagnóstico y la clasificación de los casos reportados en el presente texto 1,6.
EPIDEMIOLOGÍA
Al igual que el síndrome de Asperger, el autismo, la psicosis desintegrativa infantil y los trastornos generalizados del desarrollo no especificados, el RTT se incluye dentro de los trastornos del espectro autista, cuya frecuencia se ha incrementado entre cuatro y cinco veces durante los últimos años, relacionándose, aunque sin evidencia confirmatoria, causas genéticas, exposiciones ambientales y mayor sensibilidad diagnóstica por parte del cuerpo médico 18,29. En el 2013 se diagnosticaron en el mundo más niños con estos trastornos que con síndrome de inmunodeficiencia adquirida, cáncer y diabetes sumados 18,29.
Aunque la prevalencia global de los trastornos del espectro autista es de 620:100.000 niños 30, algunos reportes manifiestan que puede ser tan frecuente como 21:1000, afectando sobre todo a los varones en razón de 4-5:131,32. Por su parte, el RTT representa solo el 1 % de estos trastornos y afecta casi exclusivamente a las mujeres 31. En un estudio piloto realizado en 2015 con toda la población escolarizada de Quito, Ecuador, se efectuó una búsqueda activa de niños con trastornos del espectro autista, concluyendo una prevalencia local del 0,11 %, con ningún caso sospechoso ni diagnosticado de RTT 33.
La ataxia, uno de los principales criterios y síntoma diferencial del RTT, es de por sí una condición rara si no está relacionada con infecciones como la malaria. Respecto a las ataxias debidas a síndromes genéticos, según una revisión sistemática publicada por la Academia Americana de Neurología 30, la mayoría de reportes provienen de Europa con una prevalencia estimada de 14,65:100.000 niños; de estos, el RTT cuenta con una prevalencia media de 3,25:100.000 niños, basándose solo en dos series publicadas durante 1985 y una en el Pacífico Occidental en 1989. Es importante mencionar que los autores no tuvieron en cuenta motores de búsqueda regionales, no se especificaron los criterios clínicos para la definición de ataxia y ninguno de los estudios seleccionados se realizó en Colombia, por lo que se requiere más evidencia epidemiológica para conocer el estado nacional de la enfermedad.
Por ahora, la literatura acuerda una prevalencia general del RTT de 1:10.000 niñas 6,20,25,26,34-37; sin embargo, esta cifra corresponde a una extrapolación de los estudios europeos y no necesariamente debe aplicarse para Colombia ni Latinoamérica, puesto que se han encontrado variaciones importantes entre los diversos estudios epidemiológicos. En las últimas dos décadas del siglo XX, se realizaron múltiples trabajos sobre prevalencia de RTT, estimándose en Suecia de 1:15.000 niñas 38, en Suiza de 1:24.600 39, en Tokio de 0,5:10.000 40, en Dakota del Norte de 1:40.760 41, en Australia de 0,72:10.000 42 y en Noruega de 2,17:10.000 43 . En el siglo XXI sólo se encontró un estudio chino en 2007 con una prevalencia estimada de 0,57:10.000 niñas 44 , y otro de 2015 en Serbia de 0,586:10.000 45. Por su parte, el Portal de Enfermedades Raras, Orphanet [Código: ORPHA778], estima en su Informe Periódico de Prevalen-cia de Enfermedades Raras 46, publicado en noviembre del 2013, una prevalencia de la forma clásica del RTT de 4:100.000 habitantes y de 2,22:100.000 habitantes para su forma atípica.
Pese a la extensa búsqueda realizada, no se encontró ningún estudio de prevalencia para RTT en Latinoamérica y ningún caso de RTT reportado en Colombia ni de familias colombianas vinculadas al IRSF. Además, solo se localizaron en Latinoamérica una serie de siete casos en Brasil en 1990 24, un caso en México en 2007 47 y otro en Venezuela en 2012 21; ninguno de ellos contó con prueba molecular.
Caracterización clínica
El RTT debe sospecharse en pacientes jóvenes de sexo femenino con impresión diagnóstica de parálisis cerebral infantil, trastorno cognitivo idiopático, síndrome convulsivo o desaceleración del crecimiento cefálico, por lo que es recomendable la búsqueda de criterios adicionales para establecer los diagnósticos diferenciales 1,8,47. No se asocian factores de riesgo ambientales ni perinatales como la exposición a teratógenos, el nacimiento pretérmino, el bajo peso al nacer ni las condiciones neonatales como ictericia, asfixia o convulsiones 6.
Dada la gran variabilidad de presentaciones clínicas reportadas en las pacientes con RTT, es necesaria su clasificación en dos formas: el RTT clásico y el RTT atípico. Los criterios requeridos para su diagnóstico y clasificación se precisan en las tablas 1 y 2 1,12. De esta manera, la presencia de criterios de apoyo no son mandatorios para establecer el diagnóstico de RTT clásico y los criterios de exclusión no aplican para el RTT atípico.
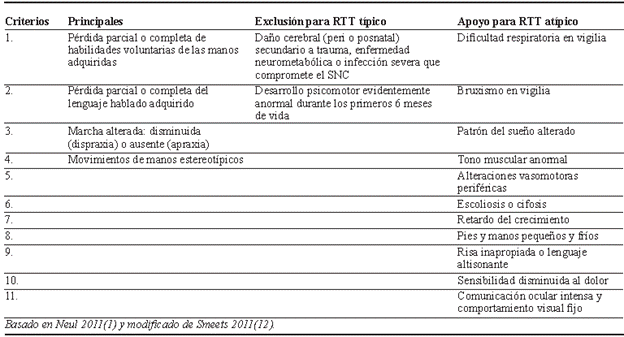

La evolución neurológica del RTT clásico ya se encuentra descrita, diferenciándose cuatro estadios tal y como se consigna en la tabla 3 6,8,12,25. Si bien estos suelen ser consecutivos, en algunos casos el deterioro psicomotor puede ser tan acelerado que pasa del estadio I al IV directamente 12. De manera adicional, el espectro fenotípico puede ser muy variado, pudiendo presentar sólo algunas de las alteraciones reportadas y cada una con diferente grado de severidad, por lo que el diagnóstico no es sencillo y los criterios de apoyo suelen ser documentados incluso en las pacientes con sospecha de RTT clásico 3,28,48.
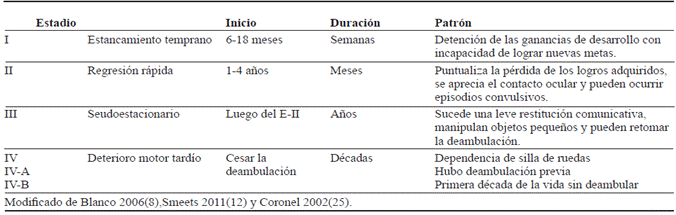
Por su parte, aunque se han descrito múltiples formas del RTT atípico 8,25,27,37, en la actualidad solo se precisan cuatro variantes: con lenguaje conservado, congénita, regresión tardía y convulsiva temprana, siendo la primera la más común 1,28 y difiriendo en pronóstico, evolución y los genes implicados 37. En la variante con lenguaje conservado, tras el estadio de regresión las pacientes consiguen pronunciar palabras aprendidas antes de la enfermedad; en la congénita no existe un período de normalidad en el desarrollo psicomotor, pero cumple con los criterios del RTT clásico; en la regresión tardía los síntomas se manifiestan de manera insidiosa a partir de los cuatro años de edad y en la convulsiva temprana comienzan los ataques antes de los seis meses de edad 8,20,37.
Dentro de los diagnósticos neurológicos diferenciales del RTT se deben considerar los demás trastornos del espectro autista 14-18,49, el síndrome de Angelman 12,50, las ataxias por otras etiologías 30 y las deficiencias comunicativas, en especial las debidas a trastornos específicos del lenguaje verbal y los trastornos globales del desarrollo psicomotor 51. Dos diferencias particulares entre el RTT y el autismo, es que las niñas con RTT mantienen la preferencia de las personas sobre los objetos y con frecuencia disfrutan de las muestras de afecto 50. El RTT diagnosticado en varones es muy raro pues cursa con encefalopatía letal temprana; los casos reportados suelen limitarse a pacientes con cariotipo 47,XXY; mutación en mosaico o a formas familiares leves, en quienes se menciona la misma evolución clínica que la presentada en mujeres 8,13,19,52.
Pese a que la descripción clásica del RTT se enfoca en las alteraciones neurológicas, algunos autores lo consideran una enfermedad multisistémica 36, dado que otros hallazgos clínicos frecuentes son: mioclonus proximal; distonía; bradicinesia; hipomimia; despertares nocturnos frecuentes; bruxismo; llanto o risa inmotivados de predominio nocturnos; episodios ansiosos; alteraciones gastrointestinales; trastornos de la deglución con disfunción orofaríngea; anoranormalidades autonómicas con cianosis periférica y problemas cardíacos como taquicardia, bradicardia sinusal o síndrome de QT prolongado; anormalidades respiratorias con apnea espontánea e hiperventilación; desaceleración en la ganancia de talla y peso; hipotrofia de las extremidades y osteoporosis por deficiencia de vitamina D 3,4,7,49,50,53,54. Además, se han relacionado algunas mutaciones del MECP2 con un incremento de los niveles del receptor de interleucina 2 y mayor población de leucocitos CD4+ con disminución de los leucocitos CD8+ y natural killer, entre otras alteraciones citohumorales, por lo que las pacientes tienen una mayor frecuencia de enfermedades autoinmunes como el síndrome de Sjögren, el lupus eritematoso sistémico, la artritis reumatoide, la esclerosis sistémica, la polimiositis y la dermatomiositis 36,48.
Las edades más peligrosas corresponden entre los 15 y los 20 años, con decesos secundarios sobre todo a neumonías, epilepsia y condiciones relacionadas con el abandono social, aunque entre el 25-33 % de las pacientes fallecen durante el sueño sin poderse establecer una causa clara, con una probabilidad de 22 % de muerte súbita durante la lactancia y en estadios avanzados de la enfermedad respecto al 2,3 % establecido para la población general 3,8,12.
Diagnóstico molecular
La etiología de RTT se ha establecido por alteraciones en el gen MECP2 localizado en la región Xq28p, cuya estructura está compuesta por cuatro exones como se evidencia en la figura 1 y codifica una proteína de unión a metil CpG 2 55. Una región de 80 aminoácidos llamada dominio de unión a metil CpG (MBD) cerca del N-terminal pareciera ser suficiente para enlazar el ADN metilado in vitro e in vivo. El MBD representa un nuevo dominio de enlace de ADN que adopta un pliegue formando una estructura en forma de cuña. También posee un dominio de represión transcripcional (TRD), que consta de 100 aminoácidos y su identificación sugiere un mecanismo activo mediante el cual MECP2 interactúa con la maquinaria de transcripción o el sistema de remodelado de la cromatina 56.
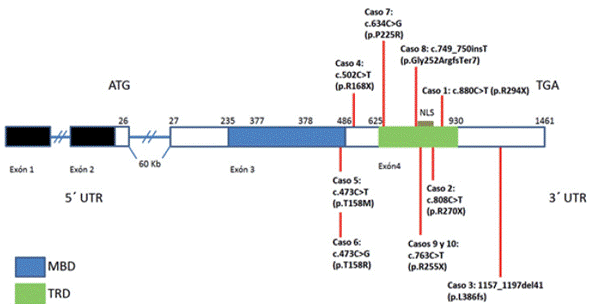
Fuente: autores
Figura 1 Estructura del gen MECP2. Se indican los dominios funcionales de unión a metil CpG (MBD), de represión transcripcional (TRD) y la señal de localización nuclear (NLS). Se señalan los sitios de las mutaciones encontradas en las pacientes reportadas.
La función de este gen se ha vinculado con la regulación de transcripción de otros genes, el desarrollo y mantenimiento sináptico, y es requerido para el aprendizaje y la memoria 57. Se sospecha que las mutaciones en el MECP2 causan una pérdida parcial o total de la habilidad para silenciar los promotores génicos que son inapropiados para tipos celulares específicos 56. Por otro lado, su sobre-expresión se relaciona con cambios en el comportamiento generando agresividad y esquizofrenia. Estudios realizados tanto en ratones knock-out como en cohortes de pacientes con fenotipos neuropsiquiátricos agresivos, encontraron polimorfismos relacionados con un aumento de función del gen o su duplicación 58. Hasta la fecha, se han reportado 960 mutaciones del MECP2 en la Human Gene Mutation Database, además de ocho mutaciones tipo cambio de sentido y truncadas, comunes para la mayoría de las pacientes con RTT en RettBASE (R106W, R133C, T158M, R168X, R255X, R270X, R294X, y R306C)52,55.
De todas maneras, se estima que el MECP2 está mutado en el 80-90 % de las pacientes con RTT clásico así como en el 40 % con RTT atípico, por lo que el diagnóstico debe ser esencialmente clínico ante la existencia de otros genes probablemente asociados, relacionándose por ejemplo, las variantes convulsiva temprana y congénita con mutaciones en los genes CDKL5y FOXG1, respectivamente 5,37,59. Con apoyo del IRSF, se publicó en 2007 la base de datos de RTT de Norteamérica con información de clasificación diagnóstica y tipos de mutaciones, y se encontró un 1,1 % de mutaciones del MECP2 sin fenotipo RTT27. Mientras tanto, en otro estudio que involucró a todos los centros genéticos de Francia, se demostró la heterogenicidad alélica del gen con una recomendación de no solo garantizar su detección sino su cuantificación 60.
No obstante y pese a la evidencia existente, todavía no se ha logrado establecer una adecuada correlación entre genotipo y fenotipo, por lo que los reportes establecen que pacientes con la misma mutación pueden presentar cuadros clínicos muy diferentes. Una explicación que se ha dado al respecto, aparte del efecto de varios genes asociados, son los perfiles de inactivación selectiva del cromosoma X que varían entre una paciente y otra, incluso dentro de una misma familia 5,55,56,61. Esto permitiría un mecanismo de transmisión por madres asintomáticas rara vez documentado, aunque el 99 % de los casos son esporádicos 52,61,62.
Manejo
Pese a todos los descubrimientos, el RTT sigue siendo un reto terapéutico para los equipos científicos en todo el mundo. Al parecer, parte de su fisiopatología incluye una sinaptopatía, por lo que podría ser manejada farmacológicamente una vez se descubran los mecanismos neurofisiológicos y moleculares exactos involucrados 3,48,59. Por ahora, los medicamentos utilizados como neuromoduladores para el control de la epilepsia y de la enfermedad de Alzheimer como la ampakina y la memantina, tienden a ser efectivos en los modelos de laboratorio y ayudan a controlar la sintomatología en las pruebas con animales 3,63. Por otro lado, mediante técnicas de silenciamiento selectivo del Mecp2 stop/y , algunos investigadores han logrado restituir el deterioro respiratorio, frenar las alteraciones comportamentales e incluso revertir los cambios morfológicos en neuronas adultas de ratones que presentaban la mutación y sintomatología de la enfermedad 3,59,64,65. Asimismo, en la página web del RSRTT, se presentan múltiples proyectos de investigación con propuestas de terapia génica, farmacológica y procedimental. Pese a todo esto, todavía no hay ningún tratamiento específico para RTT aprobado en humanos para corregir la mutación, evitar las manifestaciones clínicas o revertir el pronóstico natural de la enfermedad.
Por ahora, las medidas se centran en evitar las complicaciones médicas, mantener la funcionalidad, evitar la exclusión social y mejorar la calidad de vida de la paciente y su familia 66. Se debe brindar una adecuada asesoría genética a la familia y mantener un seguimiento interdisciplinario donde se evalúen y prevengan las comorbilidades relacionadas al RTT y las mutaciones del MECP2, como los problemas cardíacos, respiratorios, dentales, gastrointestinales, psiquiátricos, neurológicos, endocrinológicos, óseos e inmunológicos 4,7,28,36,50,53,67. Es de particular importancia el control adecuado de la epilepsia, que se presenta entre el 58 y el 90 % de las pacientes 10,28,63,68, con crisis sobretodo de tipo parciales complejas, de difícil manejo 13,49,53,63 y con tasas de control inferiores al 50% 68. Además del diagnóstico diferencial de las enfermedades autoinmunes 36, los trastornos de alimentación (que se consideran el principal punto de impacto de la calidad de vida de los cuidadores 66) y la prevención de fracturas mediante el estímulo de la actividad física, junto al suministro de multivitamínicos para suplir la deficiencia de vitamina D 4,53.
Algunas recomendaciones adicionales son el uso de aceite mineral para evitar la constipación, la asesoría nutricional para favorecer el peso y crecimiento, y enseñar las medidas de higiene del sueño para evitar las perturbaciones durante el descanso. Otros tratamientos como musicoterapia, hidroterapia, hipoterapia, psicología conductual, ortesis preventiva, ejercicio aeróbico y estimulación auditiva con música clásica en alta frecuencia han sido propuestos; sin embargo, en la actualidad no hay evidencia cientifica para su recomendación sistemática, por tanto no es posible considerarlas dentro del manejo de pacientes con síndrome de RTT 13,23,25,28,31,54,69,70.
PRESENTACIÓN DE LOS CASOS CLÍNICOS
Caso clínico 1
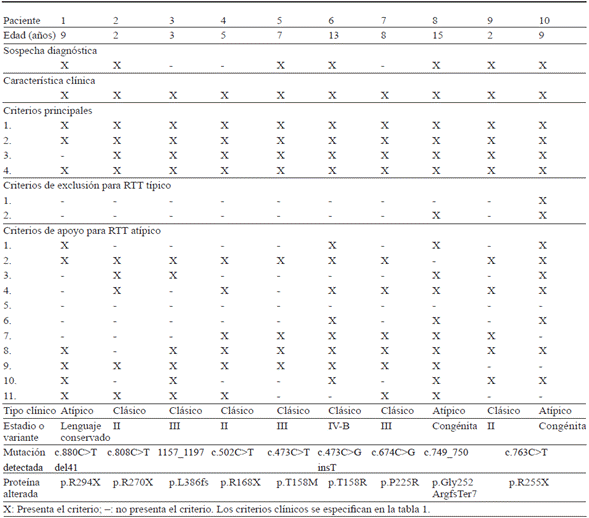
Paciente de nueve años, remitida de psiquiatría infantil y pediatría por sospecha de RTT. Producto de primera gestación, con adecuados controles prenatales, sin antecedentes personales ni familiares de importancia. Fue obtenida por parto eutócico, no se precisa peso al nacer, talla al nacer ni perímetro cefálico. No hay consanguinidad parental y tiene inmunizaciones completas. Presentó un desarrollo psicomotor normal durante el primer año: sostén cefálico a los tres meses; sedestación a los seis; pronunciación de palabras sencillas a los siete y bipedestación a los 11 meses. Posterior a los 12 meses se evidenció pérdida progresiva y completa del lenguaje asociado a hiperactividad, por lo que se estableció por parte de psiquiatría infantil un cuadro de regresión neurológica, adicionalmente movimientos estereotipados de manos, sin alteraciones de la marcha. No tenía control de esfínteres. Al interrogatorio se presentan otros criterios de apoyo para RTT (tablas 1, 2, 3, 4). Al examen físico se evidencia microcefalia (perímetro cefálico: 50 cm que corresponde a -2DS). Estaba en tratamiento con lamotrigina (Lamictal®). El servicio de genética estableció un diagnóstico clínico de RTT atípico, por lo que se realizó secuenciación del gen MECP2 con detección de mutación c.880C>T (p.R294X).
Caso clínico 2
Paciente de dos años, remitida de neuropediatría por síndrome hipotónico. Producto de primera gestación, con adecuados controles prenatales, sin antecedentes personales ni familiares de importancia. No tenía consanguinidad parental y las inmunizaciones completas. Su desarrollo psicomotor durante los primeros seis meses: sonrisa social a los dos meses; sostén cefálico a los tres; rolo a los seis; sedestación y pronunciación de palabras sencillas a los doce meses; sin gateo, bipedestación, marcha ni control de esfínteres. Posterior al año de edad, presentó pérdida del lenguaje aprendido. Al examen físico con microcefalia (perímetro cefálico: 45 cm siendo menor a -2DS), bruxismo, hipotonía, movimientos estereotipados de las manos y pérdida total del lenguaje adquirido. La cromatografía de aminoácidos fue negativa y exámenes de rutina generales normales. El servicio de genética estableció un diagnóstico clínico de RTT clásico estadio II (tablas 1, 2, 3, 4), que es complementado con prueba molecular para MECP2 con detección de mutación c.808C>T (p.R270X).
Caso clínico 3
Paciente de tres años, remitida de neuropediatría por regresión neurológica, dado principalmente por pérdida del lenguaje adquirido. Producto de tercera gestación, con adecuados controles prenatales, la madre presentó episodios febriles idiopáticos sin complicaciones durante las primeras ocho semanas de gestación, sin otras alteraciones peri o neonatales. Con diagnósticos adicionales de insomnio y asma sin tratamiento, venía en manejo crónico para epilepsia con ácido valproico y para la hiperactividad estaba siendo tratada con risperidona. Tenía inmunizaciones completas, un primo paterno con epilepsia y consanguinidad familiar. Tuvo un desarrollo psicomotor reportado como normal durante el primer año, comenzando a presentar comportamientos de agresividad y autoagresión a los 12 meses; hasta los 18 meses pronunció bisílabos, con pérdida posterior del lenguaje; marcha independiente desde los 24 meses hasta la fecha y no desarrolló control de esfínteres. Presentó adicionalmente criterios de apoyo para RTT como se describe en las tablas 1, 2, 3 y 4. Al examen físico sólo llamaba la atención la presencia de movimientos estereotipados de las manos. Tenía una resonancia magnética cerebral normal, un electroencefalograma con paroxismos de puntas-polipuntas y ondas lentas de alto voltaje generalizándose en áreas fronto-centro-parietales-temporales asincrónicas y una cromatografía de aminoácidos negativa. El servicio de genética estableció un diagnóstico clínico de RTT clásico estadio III, que se complementó con prueba molecular para MECP2 con detección de mutación 1157_1197del41 (p.L386fs).
Caso clínico 4
Paciente de cinco años, remitida de neuropediatría por sospecha de RTT. Producto de primera gestación, con adecuados controles prenatales, sin antecedentes personales ni familiares de importancia. No tenía antecedente de consanguinidad parental y tenía inmunizaciones completas. Tuvo un desarrollo psicomotor reportado como normal durante los primeros 14 meses, pero pronunciando bisílabos a los 12 meses; se detectaron movimientos inmotivados de las manos y pérdida de respuesta al llamado a los 14 meses; logró la bipedestación sin apoyo a los 19 meses y caminó a los 25 meses con dificultad para subir y bajar escaleras. No ha desarrollado control de esfínteres. Se realizó estudio auditivo, potenciales evocados auditivos que fue normal, descartando alteración de esta vía, por lo que la ausencia al llamado responde a una falla en la interacción social. Al examen físico con bruxismo, pérdida de habilidades voluntarias de las manos con movimientos estereotipados, ausencia de lenguaje verbal, hipotonía, retardo del crecimiento, pies y manos frías, risa inapropiada y establece contacto visual con fijación de la mirada, pero que pierde rápidamente. Tenía una función tiroidea conservada. El servicio de genética estableció un diagnóstico clínico de RTT clásico estadio II (tablas 1, 2, 3, 4), que es complementado con prueba molecular para MECP2 con detección de mutación c.502C>T (p.R168X).
Caso clínico 5
Paciente de siete años en control por retardo del neuro-desarrollo. No tenía antecedentes peri o neonatales, personales ni familiares de importancia, salvo por consanguinidad parental. Al examen físico llamaba la atención microcefalia (perímetro cefálico: 47 cm siendo menor a -2DS), incisivos prominentes, bruxismo en vigilia, no obediencia de órdenes, pobre seguimiento visual, movimientos estereotipados de manos y ausencia de lenguaje verbal (tablas 1, 2, 3, 4). Tenía cariotipo bandeo G de alta resolución normal. El servicio de genética estableció un diagnóstico clínico de RTT clásico estadio III, que se complementó con prueba molecular para MECP2 con detección de una variante c.473C>T (p.T158M).
Caso clínico 6
Paciente de 13 años en control por regresión neurológica y epilepsia. No tenía antecedentes peri o neonatales, personales ni familiares de importancia. Al examen físico se evidenció baja talla y peso, microcefalia (perímetro cefálico: 49,5 cm siendo menor a -2DS), sobre plegamiento del hélix, paladar estrecho y alto con mala oclusión, hipotonía, movimientos estereotipados de manos y clinodactilia del quinto dedo bilateral (tablas 1, 2, 3, 4). Tenía un cariotipo bandeo G de alta resolución normal. El servicio de genética estableció un diagnóstico clínico de RTT estadio IV-B, que se confirmó con análisis de secuenciación del gen MECP2 con reporte c.473C>G (p.T158R).
Caso clínico 7
Paciente de ocho años en control por retraso del neuro-desarrollo. Producto de primera gestación, la madre recibió un mes de progesterona oral por amenaza de aborto, con adecuados controles prenatales sin otra complicación. No tenía antecedentes familiares de importancia ni consanguinidad parental y las inmunizaciones completas. Se documentó constipación crónica, enfermedad de reflujo gastroesofágico desde el primer mes asociado a limitación para la ganancia de peso y a los dos años sufrió un episodio convulsivo atónico con supraversion de la mirada de pocos segundos de duración, manejada desde entonces con ácido valproico. Su desarrollo psicomotor: sonrisa social a los dos; sostén cefálico a los tres; rolo a los seis; sedestación con hipotonía incipiente a los siete; limitación para el gateo desde los 10; sedestación a los 12; gateo a los 18 y bipedestación a los 19 meses. A los tres años comenzó a presentar bruxismo nocturno y apneas desde el punto de vista clínico, no se había realizado polisomnografía. No había pronunciado su primera palabra ni desarrollado control de esfínteres. Al examen físico con baja talla, microcefalia (perímetro cefálico: 49 cm que corresponde a -2DS), protrusión lingual ocasional, mala oclusión, establece contacto con examinador, estado de ánimo ansioso con movimientos estereotipados de manos y realiza sonidos guturales (tablas 1, 2, 3, 4). Tenía potenciales auditivos evocados normales, electroencefalograma con focos epileptogénicos no especificados, tomografia de cráneo simple y perfiles: hematológico, hepatorrenal, inmunológico-alergénico, electrolítico, metabólico y endocrino normales. No responde adecuadamente a las terapias de neurodesarrollo. El servicio de genética estableció un diagnóstico clínico de RTT clásico estadio III, que es complementado con prueba molecular para MECP2 con reporte de c.674C>G (p.P225R).
Caso clínico 8
Paciente de 15 años, remitida de neurología clínica por retardo en el neurodesarrollo. Producto de segunda gestación, con adecuados controles prenatales, sin alteraciones peri o neonatales de importancia. Tenía una hermana con hidrocefalia manejada con válvula de derivación ventrículo-peritoneal, otra con histiocitosis, un hermano sano y tres primos del padre con retardo del neurodesarrollo. No tenía consanguinidad parental y las inmunizaciones completas. Desde los cuatro años de edad presenta epilepsia manejada con topiramato, levetiraceptam (Keppra®) y lamotrigina (Lamictal®), con un promedio de tres episodios diarios, caracterizados por movimientos tónico-clónicos generalizados, con supraversion de la mirada. Además, ha requerido cirugías por reflujo gastroesofágico y malrotación intestinal, sin otro antecedente de importancia. Con desarrollo psicomotor referido como anormal y un proceso de regresión neurológica: sonrisa social a los dos meses; sostén cefálico y rolo a los 12; sedestación a los 14; bipedestación a los 18 y primera palabra a los dos años. No ha desarrollado gateo ni control de esfínteres. Posteriormente pérdida del lenguaje y regresión motora dado por incapacidad de movilizar miembros inferiores. Al examen físico con baja talla, microcefalia (perímetro cefálico: 48,5 cm siendo menor a -2DS), escoliosis dorso lumbar, contacto visual positivo, movimiento estereotipados de manos, ausencia de lenguaje verbal, hipotrofia de las cuatro extremidades y pie izquierdo en equino varo (tablas 1, 2, 3, 4). Tenía estudios para descartar errores innatos del metabolismo con resultados normales. El servicio de genética estableció un diagnóstico clínico de RTT atípico variante congénita, que es complementado con prueba molecular para MECP2 con detección de la variante c.749_750insT (p.Gly252ArgfsTer7).
Caso clínico 9
Paciente de dos años, remitida de neuropediatría por dispraxia manual y trastorno generalizado del desarrollo. Producto de primera gestación de madre hipotiroidea controlada, sin otro antecedente perinatal, personal o familiar de importancia. No hay consanguinidad parental y tiene inmunizaciones completas. Tuvo un desarrollo psicomotor durante el primer semestre aparentemente normal, con sedestación hasta los nueve meses, bipedestación y bisílabos a los 12, logrando la deambulación sin apoyo a los 21 meses. No ha controlado esfínteres. Al examen físico se detecta microcefalia (perímetro cefálico: 43,6 cm siendo menor a -2DS), leve hipotonía, movimientos de manos estereotipados, aumento del ángulo de sustentación y ausencia de lenguaje verbal (tablas 1, 2, 3, 4). Está en seguimiento por oftalmología por hipermetropía fisiológica y por ortopedia por pie plano. Posee tomografía y resonancia magnética cerebrales, potenciales auditivos evocados y cariotipo bandeo G normales, con electroencefalograma ligeramente anormal por paroxismos epileptiformes punta onda aguda región fronto-temporal. El servicio de genética estableció una sospecha diagnóstica de RTT clásico estadio II, que se confirmó con prueba molecular para MECP2 con reporte de variante sin sentido c.763C>T (p.Arg255X).
Caso clínico 10
Paciente de nueve años, remitida de neuropediatría por retardo en el desarrollo de origen central y diagnósticos de asfixia perinatal, cuadriparesia espástica y epilepsia. Es producto de primera gestación con controles prenatales adecuados y normales, parto eutócico en semana 37 con cianosis posparto que requirió reanimación, pero no suplencia de oxígeno ni hospitalización. No desarrolló ictericia ni otras enfermedades neonatales. A los ocho meses fue hospitalizado durante tres días por constipación, sin presentar antecedentes familiares de importancia. Tiene consanguinidad parental e inmunizaciones completas. El desarrollo neuromotor fue anormal por no logro total del sostén cefálico; rolo a los cinco meses; sílabas a los ocho meses y sin sedestación ni control de esfínteres. Inició terapias físicas y de lenguaje a los 10 meses sin mención de mejoría evidente. Al año de edad comenzó a presentar crisis tónicas con reversión de la mirada por cinco minutos manejadas por neuropediatría. Fue valorada por primera vez por genética clínica a los cuatro años, donde se documentó baja talla, microcefalia (perímetro cefálico: 48 cm siendo menor a -2DS), heterocromía del iris, cuadriparesia con hipertonía, hiperreflexia en las cuatro extremidades, hipertricosis y producción de 6-7 palabras, además con hemograma, función hepática, tomografía y resonancia nuclear cerebrales, potenciales auditivos evocados, pruebas de tamizaje metabólico y cariotipo normales, por lo que el examen clínico y una búsqueda bibliográfica extensa no permitieron identificar un síndrome particular dada la probable etiología multi-factorial, manteniéndose con los diagnósticos de trabajo y el seguimiento multidisciplinario.
Durante el año siguiente presentó crisis convulsivas refractarias de difícil control, episodios de dificultad respiratoria y necesidad de gastrostomía por no alimentación, con requerimiento de enfermera domiciliaria permanente y terapias de neurodesarrollo tres veces por semana. Nuevas imágenes cerebrales (tomografía y resonancia) resultaron normales, un electroencefalograma demostró un trazado de sueño severamente anormal por ausencia de ritmos de fondo esperados, brotes frecuentes de ondas agudas y actividad lenta de alto voltaje con características epileptiformes de ubicación posterior bilateral, y la polisomnografía fue negativa para apneas/hipopneas. El nuevo examen físico describe persistencia de índices antropométricos alterados, oligodoncia por resección de piezas en mal estado, cuadriparesia espástica con temblor distal fino, buen contacto con el examinador y capacidad de producción sólo de la palabra mamá, sin sostén cefálico ni control de esfínteres. Se incluyó seguimiento por oftalmología pediátrica quien detectó miopía con astigmatismo, y fisiatría para adaptación a la silla de ruedas.
Durante el siguiente año presentó un promedio de cuatro episodios convulsivos al mes y problemas respiratorios a repetición, detectándose rotoescoliosis de vértice izquierdo con olor apocrino en las axilas y vello genital fino y rizado, por lo que se sospechó de pubertad precoz versus hiperplasia adrenal congénita. Una nueva valoración evidenció la presencia de bruxismo e identificó un perfil tiroideo normal, pero con carpograma de edad ósea/cronológica de 5:7 años y endocrino que confirmó pubertad precoz. A su vez, la radiografía de columna evidenció rotoescoliosis dorso lumbar con ángulo de curvatura izquierdo de 34 y la de cadera un índice acetabular izquierdo superior para la edad con coxa valga bilateral, por lo que se solicitaron terapias integrales de desarrollo diarias y consultas por endocrinología y ortopedia pediátricas.
A los ocho años presentó episodios convulsivos generalizados de ocho minutos de duración, se le realizó un electrocardiograma y ecocardiograma doppler con reportes normales y un estudio de hibridación genómica comparativa array (aCGH) resultó positivo para deleción del 14q21.1, sin correlación clínica pues también se detectó en la madre, quien no posee las características fenotípicas de la paciente. Se describe también tanner mamario y púbico III-IV con pie en varo bilateral, sin cambios en índices antropométricos ni otros hallazgos físicos o neurológicos de importancia. Una nueva revisión bibliográfica realizada teniendo en cuenta toda la historia clínica descrita (tablas 1, 2, 3, 4), permitió establecer una sospecha clínica de RTT atípico variante congénita, que es confirmado a sus nueve años mediante un análisis de secuenciación del gen MECP2 con reporte de mutación c.763C>T(p.Arg255X).
DISCUSIÓN
El RTT es una enfermedad compleja y de difícil diagnóstico debido a la gran variedad de manifestaciones clínicas que puede generar y su rara presentación en la población. Ya que no se encontraron caracterizaciones moleculares en Latinoamérica y las series de casos reportadas en la literatura internacional suelen poseer pocas participantes y diferentes mutaciones, no es posible confirmar la relación directa de una mutación con determinada presentación clínica 5,71,72. Teniendo eso en cuenta, se resumen las características de las pacientes y sus diagnósticos moleculares en la tabla 4 y se comparan con las descritas por mutaciones en las bases de datos de LOVD, RettBASE, Francia y Estados Unidos 27,60.
La mutación de la paciente 1 está reportada como patogénica y representa la sexta en frecuencia, siendo una mutación sin sentido que genera sobre todo formas clásicas de la enfermedad, por lo que son raros los reportes de formas atípicas, como en el presente caso. Respecto a la paciente 2, un estudio por mutaciones con RTT 72, encontró un mayor compromiso de la esfera cognitiva con esterotipias y sin distonías, representando la cuarta en frecuencia y con predominio de formas clásicas. La mutación del caso 3 se relaciona con dificultad respiratoria 71, es la onceava en frecuencia y presenta una gran variabilidad fenotípica tanto para su forma clásica como atípica, con varios reportes que la relacionan con la variedad de lenguaje conservado, aunque el presente caso tiene una presentación clásica con pérdida del lenguaje.
Las mutaciones de las pacientes 4 y 5 representan la segunda y primera en frecuencia respectivamente, con un comportamiento tanto clásico como atípico, pero sin características particulares adicionales. En el caso 6 se encontró una mutación rara que sólo ha sido reportada en una paciente de Japón según la búsqueda realizada y donde se presentó con una forma clásica leve 73. Por su parte, la mutación de la paciente 7 se ha reportado en 20 casos, con presentación tanto clásica como atípica. El caso 8 tiene una variante no reportada en la literatura, pero que es similar a la variante c.748_749insT, descrita como patogénica, teniendo ambas el mismo efecto de producir un codón stop prematuro en el codón 258 y por tanto dar lugar a una proteína truncada 60. Los casos 9 y 10 poseen la misma mutación pero con una presentación y severidad clínica totalmente distinta, lo que sugiere posibles genes adicionales asociados 56.
La epilepsia es una comorbilidad común en las pacientes con RTT, encontrándose en siete casos de la presente serie. Por su parte, el bruxismo, la frialdad distal y la risa inapropiada o lenguaje altisonante, fueron los criterios de apoyo más comunes. Pese que casi todas poseen mutaciones distintas, ninguna paciente presentó exclusivamente los criterios principales para su diagnóstico. Teniendo en cuenta lo anterior mencionado, se puede afirmar que la correlación genotipo-fenotipo es muy difícil de establecer, al menos por ahora, por lo que no es posible definir el pronóstico según el diagnóstico molecular.
Por último, se resalta la importancia de que el médico tenga claros los criterios de sospecha y diagnóstico, reevaluando el estado físico y el neurodesarrollo de su paciente en cada oportunidad de consulta. Aunque hay casos que presentan con una historia natural característica del RTT, las variantes clínicas, los factores externos y las comorbilidades presentadas, comunes en la práctica médica, suelen confundir al personal tratante y retrasar el diagnóstico. Además, dada la ausencia de reportes locales, se insta la ejecución de estudios epidemiológicos con participación de las instituciones académicas, asistenciales y científicas de la región, para poder crear un registro representativo en el que se caracterice el RTT en la población colombiana. Todo esto con el fin de mejorar la capacidad diagnóstica médica y conocer el estado actual de la enfermedad en la comunidad. La necesidad de un diagnóstico temprano radica en la importancia de la asesoría genética precoz, así como en la oportunidad de garantizar a cada paciente intervenciones específicas, preventivas y oportunas, el control multidisciplinario requerido, la inclusión en programas de apoyo sociales y una mejor calidad de vida para toda la familia.

