











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
Los aportes más representativos que se han realizado en el estudio de la genética en las epilepsias idiopáticas generalizadas, hoy denominadas epilepsias genéticas generalizadas (EGG) 1,2, provienen de estudios de agregación familiar y estudios con gemelos monocigóticos y dicigóticos -revisado en 3-. Dichos estudios sugieren que las EGG presentan un componente hereditario fuerte y determinante 4. Entre los principales síndromes epilépticos de este grupo se encuentran las epilepsias de ausencias infantiles -childhood absence epilepsy (CAE)-, juveniles --juvenile absence epilepsy (JAE)- y la epilepsia mioclónica juvenil --juvenile myoclonic epilepsy (JME)-. Una pequeña proporción de estos síndromes puede corresponder a la categoría de monogénicos y seguir modos de herencia mendeliana simple 5. En estos, una sola mutación en un gen determinado es suficiente para causar el fenotipo -revisado en 6-. La gran mayoría de estos síndromes obedece a un patrón poligénico y presenta herencia compleja, en la cual el fenotipo está determinado por una combinación de alelos de susceptibilidad distribuidos en uno o más genes y que interactúan con factores ambientales -revisado en 6-.
El estudio de las EGG comunes (las de herencia compleja) se dificulta debido a que en estas familias no se observa una relación clara entre el defecto genético y el fenotipo. Adicionalmente, factores como la heterogeneidad genética, la penetrancia incompleta y la expresividad variable contribuyen a la complejidad de las EGG comunes y complican su estudio 7.
A pesar de los anteriores factores, se ha logrado identificar varios genes relacionados con formas particulares de epilepsia. Así, mediante análisis de ligamiento y estudios de asociación se han identificado seis loci (ECA1-ECA6) relacionados con CAE. El primer locus ECA1 se superpone con el primer locus para IGE (IGE1) y ocupa el mismo lugar genético, 8q24. En esta región no se ha aislado aún el gen responsable de la variedad de fenotipos ligados a dicho locus8,9. En el caso de ECA2, se estableció que el gen que codifica para la subunidad γ-2 del receptor GABA (GABRG2) presenta mutaciones asociadas con CAE 10,11. De igual forma, en el locusECA3 (3q23) que codifica para el canal de calcio Clc-2 se han identificado seis mutaciones asociadas con diferentes subtipos de EGG 12. Asimismo, los estudios funcionales de estas mutaciones han evidenciado una alteración en la cinética de dichos canales 13-15.
Aunque se ha hecho mucho progreso en la identificación de los genes relacionados con JME, solo en tres de ellos se han identificado variantes que segregan en familias con la enfermedad. Entre ellos se encuentran el gen GABRA1, que se localiza en la región cromosómica 5q34 (JME5) y codifica para la subunidad α1 del receptor GABAA 16; el gen CLCN2, ubicado en la región 3q26 JME6/ECA3), que codifica para el canal de calcio Clc-2, mencionado anteriormente 13; y el gen EFHC1 en la región 6p12 (JME1), el cual codifica para una proteína denominada mioclonina que contiene un dominio de unión a calcio 17. Es de resaltar que las familias con frecuencia máxima de mutaciones en el gen EFHC1 son de origen hispano 17,18. Los genes restantes presentan polimorfismos asociados con la susceptibilidad a JME 19,20. Adicionalmente, se han descrito al menos otros 10 loci asociados con el síndrome, en los cuales aún no se ha identificado un gen asociado con la enfermedad 3.
Ninguno de los estudios anteriores incluye familias colombianas con epilepsia de ausencias o epilepsia mioclónica juvenil en las formas comunes. No obstante, en el caso de la epilepsia generalizada con gran mal del despertar (EGMA) se ha reportado una extensa familia de origen antioqueño 21,22 con resultados negativos. Nuestro grupo ha publicado otros estudios genéticos que incluyen familas colombianas y síndromes de EGG 23,24. Nuestro propósito fue analizar los cinco genes/loci más frecuentemente asociados con CAE, JAE o JME en un grupo de familias con estos tres síndromes de presentación común.
MÉTODOS
En este estudio participaron pacientes con diagnóstico de CAE, JAE o JME, provenientes del Programa de Epilepsia de la Universidad de Antioquia-Medellín, la Liga Central contra la Epilepsia (LICCE-Bogotá), o de la consulta privada de neurólogos coinvestigadores del proyecto. Todos los pacientes debían tener un exámen neurológico y desarrollo normal, así como disponibilidad de sus padres para participar en el estudio. Además, cada paciente debía contar con al menos seis de sus bisabuelos con ascendencia paisa (región antioqueña). La procedencia paisa se determinó con base en una encuesta de ancestralidad que tiene en cuenta la procedencia de los padres, abuelos y bisabuelos del paciente. En la misma encuesta también se preguntó por otros familiares con epilepsia. Dicha encuesta se aplicó en una visita domiciliaria que pretendía extender la genealogía, así como indagar por otros afectados en la familia.
Se calculó el poder de la muestra usando el programa GPC 25. Este cálculo se basó en las características del síndrome y los marcadores evaluados en la bibliografía reportada.
Los marcadores polimórficos SNP (polimorfismos de un solo nucleótido) se eligieron con base en los criterios de "tagging SNP" de la población CEPH derivada del norte y occidente de Europa y residente en el estado americano de Utah, disponible en el proyecto HapMap 26. La selección de los oligos o primers para la amplificación de los marcadores genéticos, así como la selección de las enzimas para la genotipificación, se llevó a cabo mediante el uso de programas y herramientas bioinformáticas disponibles en la web, como Primer3 27 o NEBcutter V 2.0 28. Los marcadores seleccionados fueron los siguientes: rs9820367 y rs2228291 para el gen CLCN2; rs6894357 y rs4428455 para el gen GABRA1; rs11135176 y rs211037 para el gen GABRG2; rs719395 y rs614570 para el gen EFHC1; rs35353620 y rs35706211 para la región ECA1.
Durante la visita a las familias de origen antioqueño, se tomó una muestra de sangre periférica (10 ml) de los padres y del hijo afectado, previo consentimiento informado. Las muestras se conservaron con anticoagulante (EDTA) a -20° C hasta el momento de la extracción del ADN. Las muestras procedentes de Bogotá-LICCE se enviaron al laboratorio y fueron almacenadas en similares condiciones. La extracción del ADN se realizó mediante el método fenol-cloroformo.
Los marcadores genéticos tipo SNP se genotipificaron mediante una PCR (reacción en cadena de la polimerasa) acoplada a una reacción de digestión con enzima de restricción (PCR-RFLP). Para algunos SNP se usó la metodología Tetra-primer ARMS-PCR (sistema de amplificación refractario de mutaciones) o una PCR alelo específica. Las PCR se realizaron en un termociclador BioRad Dyad. Posteriormente, los productos de amplificación y digestión se visualizaron en geles de agarosa teñidos con bromuro de etidio, sometidos a una fuente de luz ultravioleta con ayuda de un transiluminador UVP.
Las inconsistencias mendelianas se evaluaron usando el programa Pedcheck v 1.1. Los SNP donde se encontraron las inconsistencias se genotipificaron nuevamente. Si después de la genotipificación las inconsistencias persistían, se eliminó el genotipo del padre que presentó la incompatibilidad con el hijo. En los casos en los cuales los dos padres presentaron incongruencias con su hijo, se eliminó el dato del trío completo para el marcador en particular. También se calcularon las frecuencias alélicas y genotípicas; además, se evaluó el supuesto de equilibrio Hardy-Weinberg para cada uno de los marcadores. Estos análisis se realizaron con la librería dgcGenetics, del paquete estadístico R 29. Las medidas de desequilibrio de ligamiento se graficaron con el programa Haploview v 4.2 30. Todos los valores P se consideraron significativos si P < 0,005 (0,05/10 marcadores analizados).
Para determinar si existían diferencias genéticas entre los dos grupos de familias con JME (provenientes de Antioquia y de Bogotá) se realizaron diferentes análisis de diferenciación poblacional, como pruebas de homogeneidad alélica y genotípica, e índices de fijación, tanto en los padres como en los hijos. Estos análisis se llevaron a cabo en los programas GENEPOP 4.0.10 31 y Arlequin 3.5.1.2 32.
Se realizó un análisis de asociación alélica no paramétrica, específicamente la prueba TDT (prueba de desequilibrio en la transmisión) usando el programa Unphased 3.1.4 33. Adicionalmente, se llevó a cabo un análisis estratificado que consistió en realizar la TDT subdividiendo la muestra de acuerdo con las características clínicas del grupo de pacientes (por ejemplo sexo, edad de inicio, frecuencia de las crisis), tanto para las familias con CAE/JAE como para aquellas con JME.
Finalmente, se llevó a cabo un análisis de interacción gen-gen entre los genes evaluados.
RESULTADOS
La muestra estuvo conformada por 41 tríos familiares (caso índice y sus dos padres) y 10 dúos (caso índice y uno de sus padres) con CAE/JAE de origen antioqueño. En cuanto a JME, se incluyeron 26 tríos y 5 dúos familiares de origen antioqueño, más 16 tríos familiares procedentes de Bogotá (LICCE).
Los resultados los hemos dividido en tres secciones. Estas hacen referencia a los análisis para epilepsia de ausencias (CAE/JAE), epilepsia mioclónica juvenil (JME) y la combinación de estos tres síndromes, que en nuestro estudio integran EGG.
Pacientes con epilepsia de ausencias (CAE/JAE)
En nuestro estudio se incluyeron 24 niños y 27 niñas (razón de 0,9 niños/niña) con epilepsia de ausencias (24 CAE, 6 JAE). Según las crisis, los pacientes picnolépticos (>10 crisis/dia) fueron 29; los 17 restantes fueron no picnolépticos.
El porcentaje de genotipificación estuvo por encima de 90 % en la muestra total, con un intervalo entre 91,5 % y 94,1 °%. Nueve de los marcadores evaluados se encuentran en equilibrio Hardy-Weinberg. El marcador rs2228291 se desvió del equilibrio en la muestra conformada por los padres (P = 0,0003). Este marcador no fue incluido en las posteriores pruebas de asociación.
Los valores de LD (desequilibrio de ligamiento) solo fueron altos para los marcadores del locus ECA2, involucrando los SNP rs11135176 y rs211037 (D' = 0,816, figura 1).
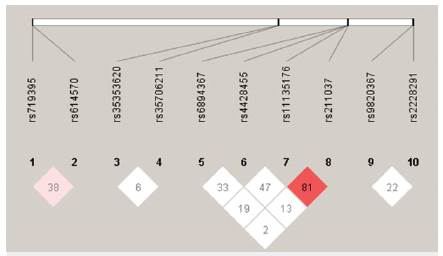
En la tabla 1 se presentan los resultados obtenidos en la prueba de desequilibrio en la transmisión (TDT), teniendo en cuenta las familias con epilepsia de ausencias, CAE/ JAE. En este análisis se encontró una fuerte asociación del fenotipo (epilepsia de ausencias) con la variante rs4428455 (P = 0,0008). Se observa que el alelo G representa riesgo. OR = 7,5, IC95 % (1,71-32,80).
Tabla 1 Resultados positivos del análisis de asociación de epilepsia genética generalizada con marcadores en los genes
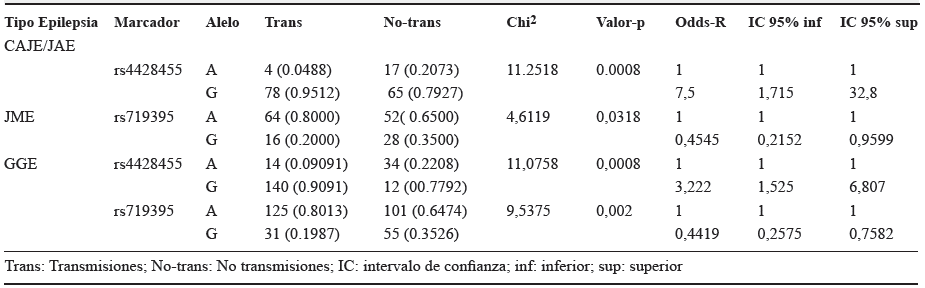
Adicionalmente, se encontraron otros valores que sugieren asociación en los marcadores rs9820367 (P = 0,03) y rs719395 (P = 0,03) con valores de OR = 0,45, IC95 % (0,220,96) y OR = 0,43, IC95 % (0,20-0,94), respectivamente. En estos dos casos, el alelo G en ambos SNP actuaría como un alelo de protección.
En el análisis teniendo en cuenta las familias con epilepsia de ausencias, en las cuales el paciente es de sexo masculino, se encontró asociación con el alelo G del marcador rs719395, con un valor de P = 0,005 y un OR = 0.21, IC95 % (0,06-0,75) (tabla 2). También se observó asociación con el SNP rs4428455 (P = 0,00004); sin embargo, el pequeño tamaño de la muestra y el bajo número de transmisiones del alelo A no permite el cálculo del valor de OR y la asociación a uno de los alelos de este locus.
Tabla 2 Resultados positivos del análisis de asociación estratificado de EGG con marcadores en los genes GABRA1 y EFHC1
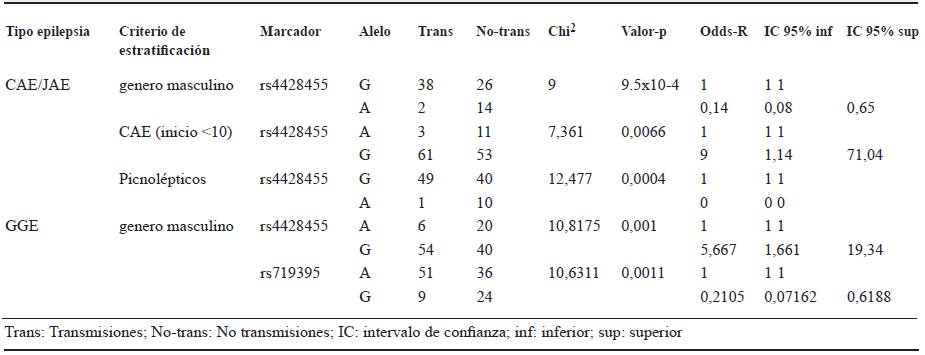
Al analizar los datos según la edad de inicio se identificó asociación del fenotipo denominado CAE (inicio < 10 años) y el marcador rs4428455, con un valor de P = 0,007 y un OR = 9, IC95 % (1,1-71) para el alelo G (tabla 2).
En los resultados del análisis de asociación en los pacientes denominados picnolépticos (pacientes con más de diez crisis por día) se encontró un valor de P significativo (P = 0,0004), que sugiere asociación al evaluar el SNP rs4428455, sin embargo, el pequeño tamaño de la muestra y el bajo número de transmisiones del alelo A impiden el cálculo del valor de OR y la asociación a uno de los alelos de este marcador (tabla 2).
Pacientes con epilepsia mioclónica juvenil (JME)
La edad promedio de diagnóstico fue 13,8 años (desviación estandar = 5,0). Los hijos afectados tuvieron una proporción 1:1 hombre: mujer.
Se encontró equilibrio de Hardy-Weinberg para todos los marcadores analizados, en ambos grupos de familias (tanto las que provienen de Antioquia como aquellas que provienen de Bogotá). El mayor LD se encontró en los SNP rs6894357 y rs4428455, con un valor de D' = 0,621 (datos no mostrados).
En la prueba de homogeneidad individual (por marcador), se observó que existen diferencias significativas en la distribución de las frecuencias alélicas en los padres (P = 0,001) y en los hijos (P = 0,006), y en las frecuencias genotípicas en los padres (P = 0,0000) y en los hijos (P = 0,0007) del marcador rs11135176 en las poblaciones evaluadas (Antioquia-Bogotá). No se observaron otras diferencias significativas (datos no mostrados).
En el análisis de homogeneidad global (teniendo en cuenta todos los marcadores) en los hijos no se observan diferencias significativas en la distribución de las frecuencias alélicas (P = 0,2164); tampoco en la distribución de las frecuencias genotípicas (P = 0,1266) entre las dos poblaciones evaluadas (datos no mostrados).
No se encontraron valores de P significativos al evaluar los índices de fijación FIS (subdivisión intrapoblacional) y FIT (endogamia total), en ninguno de los marcadores evaluados individualmente, considerando la muestra de los padres y de los hijos. Al evaluar el índice de fijación FST (subdivisión interpoblacional), se encontraron valores de P significativos en el SNP rs111335176 en los padres (P = 0,0000) y en los hijos (P = 0,001). Esto indica que hay diferenciación entre las dos poblaciones evaluadas solo en este locus. En la prueba global de diferenciación poblacional en los padres se observó un valor de FST = 0,0198 en los padres y de FST = 0,0205 en los hijos. Según estos resultados, la diferenciación entre las dos poblaciones es apenas de 2 % 34.
Con base en los resultados del análisis de diferenciación poblacional, se determinó que no existen diferencias entre las poblaciones evaluadas (Antioquia y Bogotá) en nueve de los marcadores analizados, por lo cual estas pueden considerarse como una sola población. Debido a las diferencias halladas entre las dos poblaciones en el locus rs111335176, este no se tuvo en cuenta en los posteriores análisis de asociación que consideraron el grupo de las familias de origen antioqueño y el bogotano como una sola población.
La tabla 1 muestra los resultados de los análisis de asociación en el conjunto de familias con JME. Se observó asociación del alelo G del SNP rs719395 (OR = 0,45, IC95 % 0,22-0,96, P = 0,03).
No se encontró asociación genética entre el sexo del individuo afectado con JME, ni considerando su edad de inicio, ni su frecuencia de las crisis.
Pacientes con epilepsia genética generalizada (EGG = CAE/JAE + JME).
Esta muestra se encuentra conformada por el total de las familias evaluadas en la investigación. A este grupo se le denominó familias con EGG (epilepsia genética generalizada). Se trata de los 98 pacientes y sus familiares reunidos para el estudio, grupo cuyo número asciende a 294 individuos (entre afectados y otros familiares). Según los criterios asumidos para múltiples comparaciones, dos SNP no estuvieron en equilibrio de Hardy-Weinberg. Uno de estos fue el rs2228291 y el segundo fue el rs719395. Este último se encontraba en desequilibrio solo en los casos.
El análisis de TDT en la muestra global mostró una fuerte asociación de EGG con los marcadores rs4428455 (P = 0,0008) y rs719395 (P = 0.002, tabla 1). En ambos casos, G es el alelo asociado, con un OR = 3,2, IC95 %% (1,5-6,8) y OR = 0,44, IC95 %% (0,26-0,76), respectivamente. En el primer caso, este alelo se comportaría como de riesgo, mientras que en el segundo caso actuaría como alelo de protección en el desarrollo del fenotipo EGG.
En el análisis de interacción se encontró que el marcador rs4428455 interactúa con el rs719395 (P = 0,005). Estos resultados sugieren que los individuos que portan el alelo A en rs4428455, y adicionalmente el alelo G en rs719395, presentan un riesgo superior para el desarrollo de EGG, con relación a aquellos individuos que no portan dichos alelos en estos marcadores, OR = 2,95, IC95 % (1,3-6,6) (datos no mostrados).
En el análisis de TDT estratificado teniendo en cuenta las familias en las cuales el paciente es de sexo masculino, se encontró asociación para los marcadores rs4428455 (P = 0,001, OR = 5,7, IC95 % 1,7-19,3) y el marcador rs719395 (P = 0,001, OR = 0,2, IC95 %% 0,07-0,62) (tabla 1).
En cuanto a la edad de inicio, no se encontró asociación para inicio temprano o tardío, considerando la muestra combinada para integrar EGG. Con respecto a la frecuencia de las crisis, este análisis no se realizó dado que la variable "número de crisis fue diferente en cada uno de los grupos y su medida no es equivalente.
DISCUSIÓN
El objetivo inicial de la investigación fue analizar 100 tríos familiares con EGG, distribuidos en 50 tríos familiares con epilepsia de ausencias (CAE/JAE) y 50 tríos familiares con epilepsia mioclónica juvenil (JME), todos con origen paisa. Debido a los estrictos criterios de inclusión (diagnóstico apropiado de CAE/JAE, examen neurológico y desarrollo normal, disponibilidad de ambos padres del paciente y procedencia paisa), solo fue posible completar 41 tríos y 10 dúos para las epilepsias de ausencias. En cuanto a las muestras de las familias con JME, el objetivo también consistió en la colección de 50 tríos completos; sin embargo, por los motivos antes mencionados, relacionados con los criterios de inclusión, solo fue posible colectar una muestra de 42 tríos y 5 dúos (16 de estos proceden de Bogotá-LICCE). En estas familias se evaluaron 10 SNP, dos por gen/locus candidato.
Familias con epilepsia de ausencias (CAE/JAE)
Según los análisis de calidad de los datos, solo hubo un marcador con dudosa calidad de sus genotipos. Este fue rs2228291 y por esto no se reportan más análisis para este.
En el análisis de asociación alélica, teniendo en cuenta todas las familias con CAE/JAE, se encontró una fuerte asociación del fenotipo (epilepsia de ausencias) con el marcador rs4428455 (P = 0,0008). El valor de OR = 7,5, IC95 % (1,7-32,8) indica que los individuos que portan el alelo G tienen 7,5 veces más riesgo de desarrollar CAE/JAE con respecto a los individuos que no portan este alelo.
Este marcador se encuentra ubicado en la región cromosómica 5q34, en un locus denominado ECA4 o JME5, donde se localiza el gen GABRA1, el cual codifica para la subunidad α1 del receptor GABAA. Este locus fue identificado mediante análisis de ligamiento en una familia canadiense con epilepsia mioclónica juvenil autosómica dominante (ADJME) 16, y también ha sido asociado con epilepsia de ausencias infantil (CAE) en casos esporádicos 35. Estudios funcionales indican que mutaciones en esta subunidad alteran la actividad del canal, por diversos mecanismos que van desde una disminución en la amplitud de la corriente del receptor hasta la pérdida total de la función del canal 16,35. Estos mecanismos incluyen expresión disminuida en la superficie celular de los canales que involucran alguna subunidad mutada 36, lo que ha llevado a la noción de que algunas mutaciones en estas subunidades (del receptor GABAA) podrían causar los síntomas por medio de haploinsuficiencia o dominancia negativa -revisado en 37-
Los intervalos de confianza son bastante amplios, lo cual puede deberse al pequeño tamaño de la muestra. Este comportamiento se observa también en los posteriores análisis de estratificación por sexo, edad de inicio y frecuencia de las crisis, siendo el número de individuos aún más reducido. Esto podría ajustarse aumentando el tamaño de la muestra. Para tal fin, podría incluirse pacientes de otras regiones del país, pues los criterios de inclusión agotaron los pacientes que podríamos tomar actualmente en Medellín.
Si se considera la estratificación por edad de inicio de las crisis, en los pacientes clasificados con CAE se mantuvo la asociación con el marcador rs4428455. Esto puede relacionarse quizás con un factor de severidad (edad de inicio más temprano) o con el sesgo referido a que la mayoría de la muestra CAE/JAE se compone principalmente de niños CAE.
Familias con epilepsia mioclónica juvenil (JME)
Los resultados obtenidos a partir de la estimación de los índices de fijación indican que las diferencias a nivel interpoblacional se observan principalmente en el marcador rs11135176. La prueba global en la muestra total mostró un valor de FST = 0,0261. Valores similares se encontraron al evaluar la muestra conformada por solo los padres (FST = 0,0198) y solo los hijos (FST = 0,0205). El índice de fijación Wright (FST) cuantifica la disminución en la heterocigocidad que se manifiesta en una población subdividida. Este índice tiene un rango de 0 a 1, donde 0 indica ausencia de estructura y 1 sugiere un alto grado de diferenciación entre subgrupos. Valores entre 0 y 0,05 indican poca diferenciación 34,38. Según los resultados obtenidos, la diferenciación entre las dos poblaciones, de origen antioqueño y de origen bogotano, es pequeña (0-0,05), por lo cual pueden considerarse una sola población.
La estratificación poblacional y la mezcla pueden generar resultados espurios en estudios de asociación genética, específicamente en estudios de casos y controles, por lo cual es importante que este fenómeno sea controlado o corregido de alguna manera. Una aproximación aceptable de controlar la subdivisión poblacional y la mezcla es a través de un diseño apropiado basado en familias. La prueba TDT (test de desequilibrio en la transmisión) controla la estratificación, ya que evalúa simultáneamente ligamiento y asociación 39.
El análisis de TDT en la muestra conformada por todos los pacientes con JME (Antioquia y Bogotá) encontró un valor P que sugiere asociación (P = 0,03) del fenotipo (epilepsia mioclónica juvenil) con el marcador rs719395. Sin embargo, en este caso el valor P no supera el umbral de la corrección por múltiples comparaciones (p < 0,005). El valor de OR = 0,45, IC95 % (0,22-0,96) indica que el alelo G otorgaría protección contra el desarrollo del fenotipo JME.
Diversas hipótesis se han planteado respecto al papel que desempeña este gen en la susceptibilidad para desarrollar epilepsia. Entre ellas, la más plausible podría ser aquella que involucra un efecto proapoptótico mediante la interacción con canales de calcio dependientes de voltaje. Diversos análisis funcionales plantean que las mutaciones identificadas en este gen podrían prevenir la eliminación de neuronas con una precaria homeostasis de calcio durante el desarrollo del sistema nervioso central, generando circuitos hiperexcitables susceptibles al desarrollo de crisis epilépticas 17.
Este gen es de gran importancia debido a que un número considerable de mutaciones que se han encontrado asociadas a JME fueron identificadas en familias de origen hispano (México, Honduras, Belice y Los Ángeles). En este gen se han identificado todo tipo de mutaciones en casi todos los exónes que lo conforman, con respuestas funcionales altamente variables 17,18,40. Las mutaciones más frecuentes en la población hispana son F229L, D210N, D253Y, P77T y A221H.
Sin embargo, la búsqueda de variantes polimórficas implicadas en la susceptibilidad a JME en EFHC1 no siempre ha sido exitosa. Recientemente, después de analizar una gran cohorte de familias de origen hispano en la que se hizo una búsqueda activa de variantes genéticas, este gen no resultó asociado 41. Similares resultados se encontraron al evaluar una cohorte de origen holandés 42. Se hace necesaria la búsqueda de nuevas mutaciones tanto en la región codificante del gen, así como en las regiones no codificantes, implicadas en el proceso de empalme de EFHC1 (splicing).
Los marcadores rs6894367 y rs4428455 se encuentran localizados en el gen GABRA1, el cual ha sido implicado en la susceptibilidad para desarrollar JME y CAE 16,35. El marcador rs6894357 se localiza a una distancia de 22 Kb de rs2279020, el cual ha sido asociado con resistencia a múltiples medicamentos antiepilépticos 43. Y rs4428455 se ha relacionado con epilepsia de ausencias infantil en el presente trabajo.
El análisis estratificado se realizó teniendo en cuenta la muestra completa, conformada por las familias de origen antioqueño y las de origen bogotano. En estos análisis no se encontró la asociación previamente identificada en la muestra completa. Es posible que la estratificación en este grupo de familias no haya sido exitosa en la búsqueda de asociación debido al pequeño tamaño de la muestra, así como se observó en el subgrupo de los pacientes con epilepsia de ausencias.
Familias con epilepsia genética generalizada (EGG = CAE/JAE + JME)
Se unificó la muestra de las familias con EGG. Así, se analizó un total de 98 tríos conformados por 51 familias con CAE/JAE y 47 familias con JME.
Hubo dos excepciones al supuesto de equilibrio Hardy-Weinberg. La primera fue para el marcador rs2228291 (P = 0,004) en los padres. Por esta razón, no pudo considerarse posteriormente este marcador para los análisis de asociación. La segunda excepción fue para el locus marcador rs719395 (P = 0,004) en los niños afectados. Esta observación no invalida la posterior inclusión de este marcador en las pruebas de asociación, pues la desviación del EH-W en los casos puede ser un indicativo de la asociación misma. Además, este marcador sí estaba en equilibrio en los padres, quienes se asimilan a población general.
Los análisis de asociación mostraron una fuerte participación de los marcadores rs4428455 [P = 0,0008, OR = 3,2, IC95 % (1,5-6,8)] y rs719395 [P = 0,002, OR = 0,44, IC95 % (0,26-0,76)]. En ambos casos, el alelo G fue el asociado. En el primer caso este alelo se comportaría como de riesgo y en el segundo caso actuaría como alelo de protección en el desarrollo del fenotipo EGG.
El marcador rs4428455 ya había sido identificado como asociado, en la muestra de los pacientes con CAE/JAE analizados en este estudio para el alelo G. Este marcador se localiza en el gen GABRA1, el cual ha sido asociado con epilepsia mioclónica juvenil autosómica dominante (ADJME) 16 y con epilepsia de ausencias infantil (CAE) en casos esporádicos 35. Estos dos reportes previos pueden sustentar nuestra observación consistente de la asociación de este SNP en GABRA1, pero a la vez respaldan la teoría del contínuum o de la heterogeneidad clínica de una misma variante genética.
El otro marcador asociado en este estudio, rs719395, se encuentra ubicado en el gen EFHC1, el cual codifica la proteína "mioclonina". Variantes en este gen se han relacionado con epilepsia mioclónica juvenil en familias de origen caucásico e hispanas 17. Este marcador (alelo G) también resultó asociado en el presente estudio en las familias con solo epilepsia mioclónica juvenil [(P = 0,03, OR = 0,45, IC95 % (0,22-0,96), datos no mostrados].
Debe aclararse que las variantes genéticas analizadas en este estudio corresponden a la categoría de polimorfismos y no a mutaciones. Sin embargo, estas variantes se localizan en genes previamente implicados con alguno de los síndromes de EGG. Su asociación estadística puede corresponder a uno de dos fenómenos. El primero es que corresponda a la variante funcional misma en tal gen. El segundo fenómeno posible es que la variante estudiada se encuentre físicamente cercana a otra que responde por el papel funcional. Así, la asociación identificada se debe al desequilibrio de ligamiento (LD) entre estas dos posiciones en el mismo gen.
En el análisis estratificado teniendo en cuenta el sexo, se identificó un papel causal de los dos marcadores asociados en los análisis previos (rs4428455 y rs719395), en las familias con pacientes de sexo masculino. Esta observación es llamativa, pues la relación de casos según el sexo es bastante cercana al 1:1. Es decir, este resultado no es un reflejo del muestreo preferencial por casos de un sexo en particular.
Estos marcadores se encuentran ubicad os en los loci ECA4/JME5 y JME1, donde se localizan los genes GABRA1 y EFHC1, respectivamente 16,44. En estos genes se ha reportado un gran número de mutaciones asociadas tanto a epilepsia de ausencias como a epilepsia mioclónica juvenil, principalmente en familias multigeneracionales 16-18,35,40,45,46. Sin embargo, la influencia de SNP funcionales en formas esporádicas de estos síndromes no ha sido claramente establecida 47-50.
En conclusión, nuestros hallazgos implican la participación de al menos dos de cinco genes/loci analizados. En la actualidad, estamos analizando con mayor detalle, en una muestra de familias extendidas, si las variantes analizadas acá responden directamente por el riesgo o si están en desequilibrio con otras que sí contribuyen al papel funcional para desarrollar EGG. Así, podremos determinar con mayor precisión las variantes que deberían evaluarse directamente con el propósito de establecer si el espectro clínico de las EGG realmente comparte alelos de susceptibilidad y en qué grado.

