











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
El xantoastrocitoma pleomórfico (PXA, por sus siglas en inglés) fue descrito inicialmente por Kepes y colaboradores en 1979 1; es una neoplasia astrocitaria poco común, que representa menos del 1 % de los tumores gliales 2. Suele ser prevalente en la población pediátrica y entre los adultos jóvenes hasta la tercera década de la vida. Suele tener una localización superficial que involucra la corteza y las meninges, principalmente a nivel supratentorial y en el lóbulo temporal 2, lo que hace habitual que debute con crisis convulsivas. El PXA se clasifica dentro de la categoría de otros astrocitomas, según la Organización Mundial de la Salud (OMS), y a su vez se puede categorizar como un grado II o grado III, dependiendo de la presencia de anaplasia 3. Se consideraba, convencionalmente, como una neoplasia benigna con pronóstico favorable después de la resección quirúrgica total. Sin embargo, en 1999 Giannini y colaboradores identificaron un subtipo histológico con rasgos anaplásicos (PXA-A) que muestra un comportamiento biológico más agresivo 4.
Histológicamente, el PXA presenta células pleomórficas gigantes, rodeadas por una red de reticulina, caracterizadas por la inclusión citoplasmática de vacuolas lipídicas 4-6. En contraposición, el PXA-A se diferencia por la inclusión de cuerpos eosinófilos granulares, cambios desmoplásicos, infiltración linfoide y su mayor grado de diferenciación neuronal 2,4,7.
La inmunohistoquímica es reactiva casi de manera universal para la proteína ácida fibrilar glial (PAFG) y S100 2,4,8. También se ha reportado la expresión de neurofilamentos en el 78 % de los casos, de sinaptofisina en el 40 %, de cromogranina A en el 60 %, de CD34 hasta en el 50 %, de MIB-1 (Ki67) con valores que oscilan entre el 2 y el 50 % 2,4,7-10. Recientemente, se describió la presencia de mutaciones en el oncogén BRAF (Y600E), hallazgo genotípico común de las neoplasias neurogliales (PXA, ganglioglioma y astrocitoma pilocítico extracerebeloso); este marcador puede ser útil para distinguir los PXA (el 70 % de las lesiones son positivas) de otros tumores astrocíticos difusos de bajo y alto grado, ya que este hallazgo suele ser raro en las estirpes gliales comunes 7,9,11. Por otra parte, los PXA carecen de alteraciones puntuales en la isocitrato deshidrogenasa 1 (IDH1) 8.
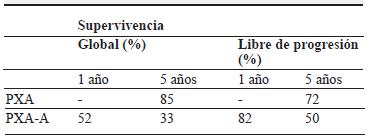
El tratamiento de elección de los PXA es la resección quirúrgica, que al ser completa permite una supervivencia del 85 y 70 % a los 5 y 10 años, respectivamente 4,12. En los PXA de bajo grado, la radioterapia y la quimioterapia no presentan ningún beneficio; no obstante, en la variante anaplásica, el uso de estas intervenciones modifica las supervivencia libre de progresión y global. En la gran mayoría de los pacientes que tienen lesiones con mayor agresividad biológica, se ha utilizado la temozolomida (TMZ) y la teleterapia, siguiendo la pauta propuesta por Stupp y colaboradores 13. Al momento de la progresión, se han empleado otros medicamentos, como las nitrosoureas, el bevacizumab (BYZ) y el vemurafenib 7,14.
A continuación, se presenta el caso de una mujer joven con el diagnóstico de un PXA-A, tratado quirúrgicamente en dos ocasiones, seguido de radioterapia y TMZ adyuvante con progresión. Después de este evento, se decidió utilizar BYZ y carmustine, logrando respuesta completa bajo criterios de Response Assesstment in Neuro-Oncology (RANO) 15.
Presentación de caso
Se trata de una mujer de 19 años de edad, quien como único antecedente familiar de importancia refirió que su abuela paterna tiene cáncer gástrico. Ella consultó por cuadro clínico que inició en el 2008, consistente en sensación de vértigo posicional, cefalea holocraneana de baja intensidad y latencia prolongada asociada a pérdida de la estabilidad sin compromiso global de la coordinación.
Fue valorada inicialmente en otra institución, donde se le realizó una TAC cerebral que documentó la presencia de un área anormal con calcificaciones. Como estudio complementario, se llevó a cabo una resonancia magnética cerebral contrastada (RM Gd), que confirmó la presencia de la lesión expansiva parietooccipital izquierda, por lo que fue programada para resección quirúrgica en octubre del 2010. La resección óptima informó la presencia de un PXA de baja gradación (ausencia de necrosis, proliferación vascular y conteo mitótico < 5 mitosis por 10 campos de gran aumento, CGA). Continuó en control imaginológico trimestral, y en noviembre del 2011 se encontró un área de encefalomalacia occipital izquierda con cambios en la intensidad de señal en T1 e hiperintensidad en T2 por compromiso nodular en el borde lateral y medial de la cavidad, debido a progresión tumoral. La espectroscopia reportó caída del pico de N-acetil aspartato (NAA) con elevación de la colina (Cho) (figura 1). En mayo del 2012, se encontró crecimiento de la lesión descrita con extensión temporal, importante modularidad y edema, además de compromiso visual. Fue reintervenida, y se documentó un PXA-A con reactividad para PFAG, Ki67 del 15 % y positividad focal para sinaptofisina. La lesión no presentaba cambios vasculares o necrosis, pero sí proliferación fibroglial focal con especial énfasis en la periferia (> 5 mitosis por 10 CGA, cobertura de 0,23 mm2). Se descartó la pérdida de 1p19q (hibridación fluorescente in situ, Yysis FISH LSI 1p36 / LSI 1q25 y LSI 19q13/19p13 Dual-Color), la expresión de olig2 y la metilación del promotor de la MGMT (PCR en tiempo real específica para la detección de la metilación de la O6-metilguanina-ADN metiltransferasa, qMSP test).
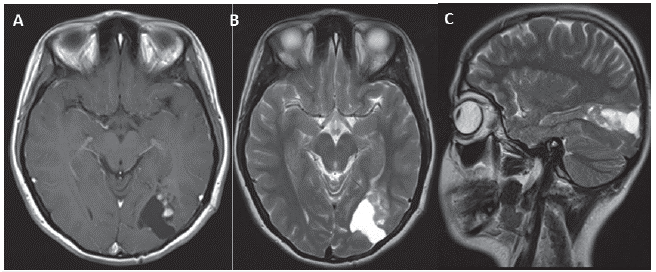
Figura 1 (A. T1 axial con contraste; B. T2 axial; C. T2 sagital). Cambios postquirúrgicos temporooccipitales con cavidad de resección con realce nodular en el borde lateral de la cavidad de resección compatible con residuo tumoral. <no hay prolongación del T2 perilesional.
Por el contrario, la genotipificación para BRAF (PCR en tiempo real alelo específica para la detección de la sustitución Y600E, Cobas 4800) demostró la alteración puntual Y600E.
En el momento de la valoración neurooncológica inicial, solo se halló la alteración campimétrica temporal derecha. Se inició tratamiento con quimiorradiación, siguiendo la plataforma propuesta por Stupp y colaboradores, completando 60 Gy (200 cGy/día) en octubre del 2012, seguidos de dos ciclos de adyuvancia con TMZ (150 y 200 mg/m2/ día por 5 días cada 28 días).
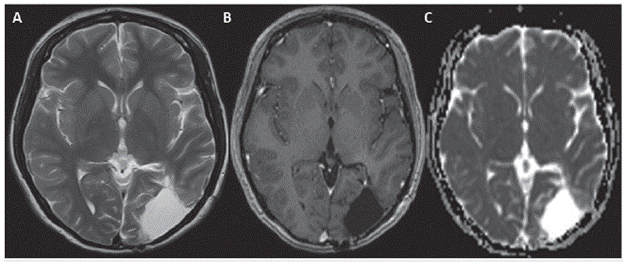
En enero del 2013, se realizó una nueva secuencia imaginológica que demostró aumento de la lesión nodular (16 x 9 mm) con elevación de la Cho y reducción del NAA, más cambios en la perfusión (T1) por aumento de la angiogénesis (figura 2). Desde entonces, presentó crisis tónico-clónicas generalizadas y afasia nominativa, hallazgos sugestivos de una nueva progresión de la enfermedad, confirmada ima-ginológicamente por RANO que demostró evolución de la lesión nodular cuatro semanas después de la progresión, aunada a marcada elevación de la Cho, alteración de la relación Cho/NAA y aumento del volumen sanguíneo cerebral relativo (rCBY). Se inició tratamiento con levetiracetam a razón de 500 mg cada 12 horas y se incrementó la dosis hasta un máximo de 1000 mg cada 12 horas. Se efectuó una resección subtotal (la histología confirmó la presencia de PXA-A) y en febrero del 2013 se inició la segunda línea con carmustine (80 mg/m2 días, 1 a 3 cada 8 semanas) y BYZ (10 mg/kg cada 15 días). Hasta el momento se han completado seis ciclos (12 meses de intervención) en respuesta completa (figura 3). Esta intervención permitió la regresión completa de las crisis y la resolución completa del trastorno del lenguaje.
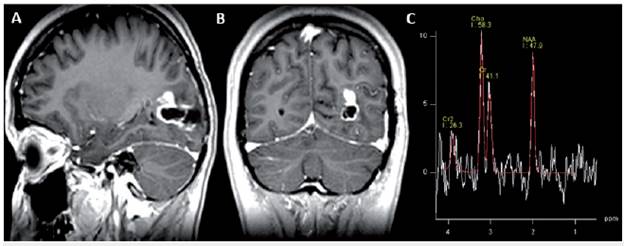
Figura 2 (A. Sagital T1C; B. Coronal T1C; C. Espectroscopia Univoxel). En la figura se observa la cavidad de resección temporooccipital izquierda con aumento en el realce nodular comparado con el control postquirúrgico, especialmente hacia el aspecto superior de la cavidad. Espectroscopía con aumento en el pico de colina y descenso del pico de N acetil-aspartato compatible con lesión tumoral.
DISCUSIÓN
En el 2004, Tekkõk y colaboradores reportaron 200 casos de PXA en un período de 25 años; un estudio posterior en el 2007 registró 40 casos adicionales 12 y Tamara M. Yu y colaboradores agregaron 84 pacientes de forma más reciente 2. A partir del análisis agrupado de estas series, se puede definir que el PXA es un tumor de pacientes jóvenes, que presenta su pico de incidencia entre los 10 y los 30 años. La edad media de los casos reportados es de 25,2 años 2,4,12,16.
Aunque la localización suele ser supratentorial en la mayoría de los casos, también se ha reportado en cerebelo, médula cervical y toracolumbar, retina, hipotálamo, tálamo, cuerpo calloso, glándula pineal, silla turca y tronco cerebral 2,4,6. Dada la ubicación hemisférica superficial de los PXA, la presentación clínica más común son las convulsiones, evento que ha sido reportado en el 70 % de los pacientes 4, tanto así que muchos de ellos tienen el antecedente de epilepsia meses o años antes del diagnóstico del tumor. Otras manifestaciones incluyen el déficit neurológico focal, alteración de la visión, cefalea y ocasionalmente hemorragias intracerebrales 2.
La presencia de células xantomatosas es patognomónica, pero no universal, y está presente en el 60-66 % de los casos 2,4,6. Los cuerpos granulares eosinofílicos son relativamente comunes, junto con los depósitos de fibras de reticulina. Las fibras de Rosenthal son menos comunes 2,4,16,17.
Las características inmunohistoquímicas distintivas de los PXA-A son el índice mitótico elevado (entre 5/50 CAP y 32/10 CAP) 7, la disminución de la positividad para marcadores de diferenciación neuronal y el aumento en el Ki67, que puede llegar hasta el 50 %. De manera adicional, poco más de la mitad de los pacientes presenta la mutación Y600E del BRAF y el 85 % carece de la metilación del promotor MGMT 7,18.
El tratamiento quirúrgico suele ser la primera elección en la mayoría de los pacientes con PXA-A; no obstante, la mediana de tiempo hasta la recurrencia oscila alrededor de los 12 meses. Una pequeña serie reportó que en los sujetos donde se logró una resección completa, el 50 % tuvo recurrencia del tumor y antes de 15 meses. En la misma dirección, el 55 % de aquellos tratados con resección parcial tuvo progresión antes del sexto mes 2. Dentro de las opciones terapéuticas complementarias para los PXA-A, Schmidt y colaboradores propusieron la de quimiorradiación con TMZ, seguida con el mismo agente en adyuvancia con este esquema; el 60 % con PXA-A tuvo una supervivencia que se extendió a los 10 años en algunos casos.
Con anterioridad, se reporta un caso tratado con cirugía radical, seguida de teleterapia parcial (suspendida por intolerancia), TMZ y, posteriormente, carmustine al momento de la recurrencia, que continúa en seguimiento después de 7,5 años. Hace poco se reportó el uso de BYZ y TMZ en un paciente con PXA-A refractario que logró supervivencia, hasta el momento del reporte de siete meses 7. Recientemente, se publicó una serie de cuatro pacientes con PXA portadores de la mutación del BRAF tratados con vemurafenib al momento de la recurrencia. A pesar del limitado tamaño muestral, los autores encontraron una respuesta favorable en el 50 % y a un mayor tiempo de supervivencia libre de progresión (SLP) mayor a seis meses 14. La tabla 1 incluye la LSP y la SG entre los PXA y los PXA-A 2,4.
Un experimento clínico fase II (BELOB) presentado en el 2013 incluyó 153 pacientes con glioblastoma recurrente, a los cuales les dieron manejo con BYZ y lomustine (Lom), en combinación e individualmente, y se halló una diferencia entre los grupos para la SG del 25 % a favor de la combinación cuando se comparó con BYZ o Lom, respectivamente. Así, se reportó una ganancia final de cuatro meses para este desenlace y una reducción en el riesgo de progresión a seis meses del 28 % debido a este esquema de tratamiento combinado. La toxicidad más prevalente fue la trombocitopenia grado 3-4 (63 %), seguida de la fatiga (18 %). Con base en estos hallazgos, se extrapoló el tratamiento para la paciente descrita en este artículo, con el cual se ha obtenido una respuesta completa 19.
Sin embargo, y a pesar de los resultados favorables en el estudio BELOB, un estudio fase III publicado recientemente por Wick y colaboradores, donde se buscó el beneficio de la terapia combinada con lomustina/bevacizumab, falló en encontrar beneficios en términos de sobrevida global. Si bien cabe resaltar que este beneficio no fue observado en la mayoría de los pacientes, es pertinente reconocer que un subgrupo de pacientes sí presentó dicho beneficio 20. Si se toman en cuenta los desenlaces antes mencionados, así como los resultados publicados en una cohorte de pacientes latinoamericanos 21, en donde se vio que existe un beneficio términos de tasas de respuesta, consideramos que quizá haya diferencias biológicas entre las poblaciones que brinden a los pacientes latinoamericanos una mejor supervivencia. Si bien estos resultados son alentadores, deberán ser validados en estudios aleatorizados donde se haga un reclutamiento más significativo de esta población.
CONCLUSIÓN
Los PXA-A, que son tumores raros, se encuentran en diferentes series de casos en los últimos años, en las cuales se ha logrado identificar las características histológicas más importantes, así como la inmunohistoquímica relevante para lograr una mejor identificación. De igual forma, hallar nuevos esquemas terapéuticos y mejorar la sobrevida de estos pacientes, como es el caso de la mutación Y600E del oncogén BRAF y el uso de vemurafenib, que podría convertirse en parte de un protocolo de manejo de estos tumores.

