











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkINTRODUCCIÓN
La esclerosis múltiple (EM) es una enfermedad inflamatoria crónica de origen autoinmune del sistema nervioso central (SNC), que conduce a daño progresivo de la mielina y el axón por medio de mecanismos de desmielinización y degeneración neuronal 1. Cerca del 85 %% de los pacientes presentarán un curso remitente-recurrente cuya primera manifestación es el síndrome clínico aislado (SCA) 2. El SCA se define como el primer episodio clínico (síntomatico) compatible con EM, pero que aún no cumple con los criterios clínicos de diseminación en tiempo ni espacio 3.
El abordaje del SCA es indispensable para establecer el riesgo de evolucionar a EM clínicamente definida (EMCD), además de guiar de forma temprana una conducta terapéutica 3. El objetivo de esta revisión es profundizar en los conceptos básicos del SCA y su relación con la EM, buscando responder a las siguientes preguntas:
- ¿Qué es el SCA y cuáles son sus manifestaciones clínicas?
- ¿Cuáles son los factores predictores de conversión a EM?
- ¿Qué diagnósticos diferenciales se deben tener en cuenta?
- ¿Se debe tratar el síndrome clínico aislado?
El síndrome clínico aislado
El SCA es un término ampliamente usado para describir el primer episodio clínico de signos y síntomas sugestivos de un proceso inflamatorio y desmielinizante en el SNC que pueda ser compatible con EM 2,3. Los síntomas del SCA deben durar por lo menos 24 horas y no estar acompañados de fiebre, procesos infecciosos o encefalopatía. El SCA, como su nombre lo indica, debe tener un curso aislado en el tiempo y el espacio (monofásico y monofocal, respectivamente).
El termino SCA tambien ha sido usado para agrupar pacientes de acuerdo con una variedad de criterios:
i) Único evento de sintomas y signos clínicos que indican una única lesión (enfermedad aislada en espacio y tiempo). SCA clásico.
ii) Episodios recurrentes en una única localización (enfermedad aislada en espacio, pero no en tiempo).
iii) Único evento clínico donde los sintomas y/o hallazgos al examen neurológico sugieren la presencia de dos o más lesiones en localizaciones diferentes (aisladas en tiempo, pero no en espacio).
Una amplia revisión de la literatura demostró que el 46 % de los pacientes debutan con síntomas sensitivos o motores, 23 °% con síntomas multifocales, 21 % con neuritis óptica y 10 °% con síntomas de tronco encefálico 4. Con menor frecuencia se observan manifestaciones difusas tales como crisis convulsiva o alteración del estado de conciencia; estas manifestaciones no se consideran de entrada como un SCA clásico y obligan a considerar un amplio estudio en busca de diagnósticos alternativos 5,6. Otras manifestaciones menos frecuentes pueden incluir síntomas paroxísticos como disartria, ataxia, o síntomas sensitivos de corta duración 7, por lo que el neurólogo debe indagar con cuidado acerca de los síntomas, su duración y distribución, con el objetivo de descartar otras patologías. Las manifestaciones atípicas en el SCA se consideran banderas rojas determinantes en el diagnóstico diferencial (figura 1).
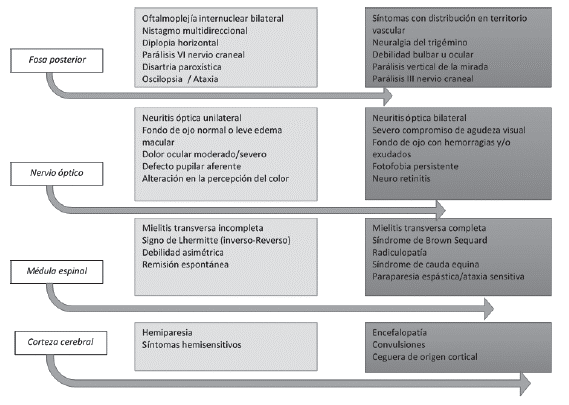
Figura 1 Manifestaciones típicas (gris claro) y atípicas (gris oscuro) del síndrome clínico aislado. Se muestran los síntomas de acuerdo con su distribución topográfica en el sistema nervioso central (SNC).
La edad de presentación del SCA es similar a la edad del diagnóstico de EM, con una media de 30 años, y la relación mujer: hombre es 2-5 a 1.
El síndrome clínico aislado se diferencia del síndrome radiológico aislado (SRA), ya que este último se caracteriza por el hallazgo incidental de lesiones hiperintensas en T2 sugestivas de un proceso desmielinizante en un paciente al que se le realizó una resonancia magnética (RM) con una indicación diferente al estudio de una enfermedad desmielinizante 8.
Manifestaciones clínicas frecuentes
- Neuritis óptica: es una de las presentaciones frecuentes del SCA. Se caracteriza por ser dolorosa, unilateral y de instauración subaguda. El paciente puede cursar con síntomas de visión borrosa, escotoma central, ocasionalmente fotopsias y/o defecto altitudinal. Habitualmente se resuelve de manera espontánea dentro de las primeras cuatro semanas 9,10. El compromiso de la agudeza visual suele ser moderado y en pocas ocasiones severo, por lo que encontrar pacientes que refieren solo percepción de la luz es extremadamente raro y obliga a descartar otras causas de compromiso del nervio óptico. Al examen de fondo de ojo se puede encontrar un discreto edema de papila, pero por lo general es normal 11; tambien se puede evidenciar un defecto pupilar aferente relativo y desaturación a la visión de colores.
- Síndrome de tronco encefálico: los pacientes que debutan con este síndrome típicamente presentarán diplopía binocular y en la exploración neurológica se podrá objetivar una oftalmoparesia internuclear o una monoparesia aislada del III, IV o VI nervio craneal. Otras manifestaciones clínicas pueden ser ataxia, disartria, dismetría, vértigo y/o síntomas sensitivos faciales.
- Síndrome medular: se presenta con síntomas de mielitis sub-aguda incompleta. Con frecuencia los pacientes refieren disminución de la fuerza o sensibilidad segmentaria y asimétrica, trastorno de esfínteres (mayor compromiso del esfínter urinario sobre el anal), banda de hipoestesia o diestesia en tórax o abdomen y signo de Lhermitte 12.
- Sindrome hemisférico: se puede manifestar como un trastorno hemimotor o hemisensitivo con compromiso simétrico de la cara y extremidades, particularmente cuando hay compromiso en el tracto corticoespinal y espinotalámico. De manera poco frecuente puede iniciar con alteraciones cognitivas (34 a un 65 °% de los pacientes), apraxias, convulsiones, afasia e incluso hemi-negligencia.
FACTORES PREDICTORES DE CONVERSIÓN A ESCLEROSIS MÚLTIPLE
Entender los factores predictores de conversión a EM en pacientes con SCA permite estratificar y evaluar el riesgo de EMCD y así guiar las desiciones terapéuticas. En la actualidad, se han estudiado diferentes factores pronósticos clínicos y paraclínicos como la RM, la presencia de bandas olicoglonales (BOC) en líquido cefalorraquídeo 13 y los potenciales evocados (PE) 14. Aún en vía de estudio se encuentran algunos biomarcadores séricos como la cadena liviana de los neurofilamentos y la chitinasa 3; sin embargo, hasta la fecha no existe ningún biomarcador exclusivo de esta enfermedad, por lo que el uso de todas las estrategias disponibles nos acercará de manera adecuada a la estratificación del riesgo y la toma de decisiones terapéuticas.
Hallazgos en resonancia magnética
Hasta el momento, la RM es la herramienta de prónostico más utilizada en el SCA 15. La presencia de lesiones desmielinizantes sugestivas de EM se convierte en un factor de riesgo importante para evolucionar a EMCD en los pacientes que se presentan con SCA 16. Estudios recientes demuestran que el 50 al 70 °% de los pacientes que se presentan con SCA tienen lesiones desmielinizantes asintomáticas en la sustancia blanca cerebral y/o medular en la RM. Tres estudios con seguimiento a 7, 15 y 20 años concluyen que existe un 65 %%, 72 %% y 80 %% de riesgo conversión a EMCD cuando se encuentran lesiones en la RM; comparado con un 8 %%, 25 %% y 20 %%, respectivamente, si la RM inicial es normal 16-18. Recientemente se ha demostrado que los pacientes con lesiones localizadas en la fosa posterior, en particular en el tronco cerebral, presentan un mayor riesgo de conversión a EM que pacientes con lesiones en otras localizaciones 19. Por lo tanto, la presencia y el número de lesiones en la RM reflejan la actividad subclínica y se convierten en predictores de conversión a EM durante los próximos 5 a 15 años. 3,20. Además de las lesiones del troco encefálico, las lesiones con mayor valor pronóstico son aquellas que realzan en forma de anillo incompleto en la sustancia blanca tras la administración de gadolinio o con realce homogéneo multifocal 21, lesiones hiperintensas en T2 que afectan al cuerpo calloso o la presencia de lesiones en el cordón posterior de la médula espinal, sobre todo la médula cervical 22.
Recientemente se ha conocido una nueva revisión propuesta para los criterios de McDonald (revisión de 2017) que incluye la presencia de BOC en el LCR en pacientes con un primer síntoma desmielinizante, de tal forma que "En un paciente con SCA típico y el cumplimiento de los criterios clínicos o de RM para diseminación en espacio y no mejor explicación para la presentación clínica, la demostración de BOC específicas en el LCR permite que el diagnóstico de EM sea hecho sin esperar por el criterio previamente requerido de diseminación en tiempo" (tabla 1) 23.
Tabla 1 Criterios de Mc Donald 2017 para esclerosis múltiple (diseminación en tiempo y espacio) en pacientes con CIS. * En un paciente con SCA típico y que cumple los criterios de diseminación en espacio; en ausencia de una mejor explicación clínica, la presencia de BOC en LCR permiten el diagnóstico de EM

Aunque la atrofia cerebral tanto de sustancia blanca como gris no se encuentra dentro de los criterios diagnósticos en EM, un estudio de seguimiento a cuatro años demostró que en pacientes con SCA la atrofia regional o global segmentada de estructuras como el tálamo, el giro frontal superior y la sustancia gris del cerebelo son predictores independientes para conversión a EM 24.
El protocolo recomendado por el Consortium of Multiple Sclerosis Centers (CMSC) 25 y Magnetic Resonance in Multiple Sclerosis (MAGNIMS) 26 para la realización de la RM cerebral en pacientes con SCA o sospecha de EM debe incluir la exploración en tres planos (3D), que incluye la secuencia FLAIR sagital y axial, T2 spin eco densidad de protones axial, T1 axial con y sin gadolinio (Gd) y difusión por resonancia magnética (DWI). Se recomienda realizar la RM medular si la RM cerebral no es suficiente para establecer el diagnóstico de EM o si el paciente exhibe manifestaciones neurológicas de cordón medular. Sin embargo, en muchos centros se realiza como parte del estudio inicial de cualquier paciente con sospecha de EM. El seguimiento por imágenes se debe realizar cada seis meses a dos años en todos los pacientes a quienes se realiza diagnóstico de esclerosis múltiple recaída remisión (EMRR).
Biomarcadores de sangre perifícerica / LCR como predictores de conversión a EM
Los biomarcadores permiten establecer el diagnóstico y las pautas terapéuticas y de pronóstico de manera temprana. La definición del término biomarcador se puede resumir en los siguientes aspectos:
- Se considera biomarcador cualquier parámetro biológico que se puede medir objetivamente, y es un indicador de procesos biológicos normales, patológicos o de respuestas a intervenciones terapéuticas específicas 27).
- Un biomarcador idealmente serviría como un "punto final sustituto" de un resultado clínico, solo si es plenamente capaz de representarlo. Sin embargo, para determinar el riesgo de conversión a EM hasta ahora no existe ningún biomarcador que cumpla esta característica.
La determinación de BOC en LCR, como reflejo de la síntesis de inmunoglobulina G en el compartimiento intratecal, es muy importante a la hora de establecer el riesgo de conversión a EMCD. Entre el 60 %% y el 70 %% de pacientes con SCA, y más del 90 %% de pacientes con EM tienen dos o más BOC en LCR al comparar con el suero (patrón tipo II) 3,28. Para la detección de las BOC es importante tener en cuenta aspectos técnicos, se deben realizar de forma simultánea en suero y LCR dentro de las primeras 72 horas de obtención de las muestras, además de utilizar el método de isoelectroenfoque con gel de agarosa seguido de inmunodetección por transferencia o fijación 29. La presencia de dos o más BOC en el LCR tiene un valor predictivo positivo del 97 %%, valor predictivo negativo de 84 %%, sensibilidad del 91 %% y especificidad del 94 %% para el riesgo de conversión a EMRR 30.
En el estudio publicado por Tintoré y colaboradores, al analizar de forma conjunta los datos de RM y el resultado de las BOC se concluyó que la presencia de BOC aumenta el riesgo de conversión de 59 %% a 64 %% a EM en un paciente con una RM anormal. En ese mismo estudio se demostró que la presencia de BOC en pacientes con SCA y estudio RM normal incrementaba el riesgo de EM de un 4 %% a 23 %% 31. Tintoré y colaboradores 31 y Masjuan y colaboradores 30 demostraron que tener BOC en LCR después de tres meses de un primer episodio clínico dobla el riesgo de un segundo brote a los 50 meses y seis años, respectivamente. Con estos resultados, podemos concluir que el riesgo de progresión a EM clínicamente definida en pacientes con RM de neuroeje normal y ausencia de BOC en LCR es bajo; mientras que este riesgo es alto en pacientes con RM con lesiones desmielinizantes y BOC presentes.
Otro biomarcador que se ha estudiado en los últimos años en la presencia de chitinasa-3 en LCR. Sus niveles se elevan durante la inflamación y se ha asociado con un mayor riesgo de conversión a EMCD. Comabella y colaboradores, a través de un análisis proteómico, compararon los niveles de chitinasa-3 al momento del SCA de 30 pacientes que tuvieron conversión a EM, con los niveles de chitinasa-3 de 30 pacientes con SCA que no progresaron a EM. En este estudio se demostró que los niveles elevados de este biomarcador al momento del SCA predicen el riesgo y el tiempo de conversión a EM 32.
Recientemente se han estudiado los niveles de la cadena liviana de neurofilamentos (componentes esenciales del citoesqueleto protéico axonal que pueden indicar daño axonal); los valores elevados se han relacionado con mayor riesgo de conversión a EM 33,34.
En la actualidad, el uso de niveles de chitinasa-3 y cadena liviana de neurofilamentos no está indicado en la práctica clínica diaria. Pero el uso de las BOC es ampliamente aceptado y debe hacer parte del estudio paraclínico inicial en todos los pacientes con SCA.
Predictores clínicos y demográficos
Algunas manifestaciones clínicas en el SCA están asociadas con un mayor riesgo de evolucionar a EMCD durante los dos primeros años de seguimiento. Dentro de ellas se encuentra el inicio de síntomas antes de los 30 años, etnia no caucásica 35, sexo femenino 36 y el compromiso de varios sistemas funcionales. Sin embargo, dos estudios recientes no encontraron asociación entre los síntomas multifocales y el incremento del riesgo de conversión a EM 37,38. En pacientes que debutan con manifestaciones monofocales, se demostró que la carga lesional (más de nueve lesiones en T2) y la presencia de actividad (por lo menos una lesión que realza con Gd) son los predictores más fuertes de conversión a EM en un tiempo más corto (HR 95 %% CI: 2,13/1,05-4,34; p = 0,032; HR 95 %% CI: 2,28/1,24-4,18; p = 0,006, respectivamente). Otros estudios no han demostrado un incremento del riegso de conversión según el sexo 39 ni el orígen étnico 40. En conclusión, los factores clínicos y demográficos no parecen ser predictores de gran peso en el riesgo de conversión a EMCD.
DIAGNÓSTICO DIFERENCIAL
Es fundamental como parte del estudio inicial de los pacientes con SCA descartar de forma razonable posibles diagnósticos alternativos, principalmente cuando las manifestaciones clínicas y radiológicas son atípicas.
Estas patologías podemos dividirlas en tres grandes categorías:
- Otras enfermedades inflamatorias desmielinizantes idiopáticas del SNC: encefalomielitis aguda diseminada (ADEM), neuritis óptica idiopática, mielitis transversa idiopática, espectro de neuromielitis óptica (ENMO).
- Enfermedades inflamatorias sistémicas con manifestaciones en SNC: neurosarcoidosis, angeítis primaria del SNC, lupus eritematoso sistémico (LES), enfermedad de Behcet, vasculitis, entre otras.
- Enfermedades no inflamatorias del SNC: patología vascular isquémica o hemorrágica, lesiones tumorales del SNC, manifestaciones neurológicas de enfermedades metabólicas, infecciones.
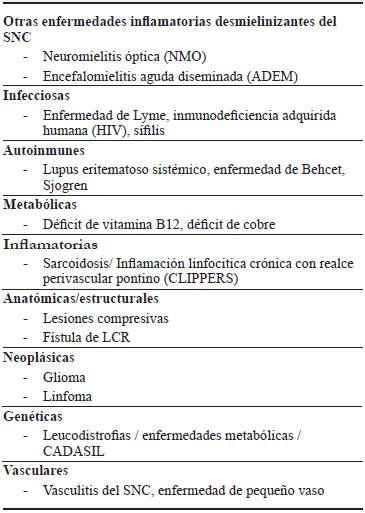
Se puede orientar con mayor certeza el diagnóstico de trabajo y excluir patologías infecciosas, inflamatorias, metabólicas, genéticas, tumorales, vasculares o autoinmunes 41 (tabla 2), dependiendo de las características étnicas, etarias, comorbilidades y manifestaciones clínicas de cada paciente. Las enfermedades del ENMO eran previamente consideradas una variante de la EM 42, sin embargo, en la actualidad es uno de los diagnósticos diferenciales a tener en cuenta, sobre todo en nuestro medio donde la prevalencia de la EM es baja.
Tabla 2 Diagnósticos diferenciales en CIS. ADEM: encefalomielitis aguda diseminada. CLIPPERS: Inflamación linfocitica crónica con realce perivascular pontino. LCR: Líquido cefalorraquídeo
El primer episodio clínico de la NMO puede ser mono o polifásico 43, siendo las manifestaciones clínicas más agresivas (compromiso de varias vías medulares y agudeza visual menor a 20/200) y con menor recuperación a pesar del tratamiento 44. Una de las características cardinales del ENMO es la presencia de una lesión medular longitudinalmente extensa (tres o más cuerpos vertebrales comprometidos) que ocupa más del 50 %% del diámetro medular, con localización de predominio central. Por otro lado, las lesiones medulares en el SCA no suelen sobrepasar dos segmentos vertebrales y tan solo un tercio del diámetro cordonal se encuentra comprometido, siendo más frecuente la afectación de cordones laterales o posteriores. Otra clave diagnóstica para ENMO son los hallazgos en la RM cerebral; en la mayoría de los casos suele ser normal, pero tambien se pueden identificar lesiones hiperintensas en el cuerpo calloso, bulbo raquídeo y órganos circumventriculares. En el LCR se puede encontrar pleocitosis neutrofílica y con muy baja frecuencia se observan BOC positivas (menos del 10 %%) 45. La detección de anticuerpos anti aquaporina 4 en suero es de gran ayuda, pues tienen una sensibilidad del 73 %% y una especificidad del 91 %%, que sumados con la aplicación de los criterios de diagnósticos del 2015 46,47 confieren certeza diagnóstica.
Por otro lado, la encefalomielitis aguda diseminada (ADEM) es más frecuente en niños y por tanto debe ser tenida en cuenta como diagnóstico diferencial, sobre todo en pacientes jóvenes con SCA que debutan con síntomas atípicos. Para establecer el diagnóstico de ADEM se requiere un compromiso multifocal del SNC (afectando nervio óptico, tronco encefálico, cerebelo, hemisferios cerebrales y/o médula), acompañado de encefalopatía, que se puede manifestar en forma de alteración comportamental, compromiso del estado de conciencia o convulsiones. Tiene un curso monofásico y generalmente se identifica un proceso infeccioso o vacunal los días o semanas previos al inicio del cuadro clínico. En cuanto a los hallazgos paraclínicos, se puede encontrar pleocitosis (< 50 leucocitos) e hiperpro-teinorraquia en el LCR, y las BOC son usualmente negativas 48. En la RM se evidencia compromiso multifocal, bilateral, asimétrico de la sustancia blanca que se puede extender a la sustancia gris cortical o profunda, con captación de contraste y edema en el 30 %% de los casos 49. En las RM de seguimiento puede haber estabilidad o incluso importante mejoría de las lesiones. Desafortunadamente, en la actualidad no existe ningún biomarcador o estudio por imagen que permita distinguir las dos entidades 50.
TRATAMIENTO
El tratamiento del SCA se puede dividir en dos fases. En la primera, se realiza el tratamiento agudo de los síntomas por los que consulta el paciente, en tanto que en la segunda, una vez estratificado el riesgo de conversión a EM clínicamente definida, tiene lugar el inicio de medicamentos modificadores de la enfermedad.
Tratamiento agudo
Aunque la gran mayoría de los eventos clínicos son leves y cursan con una recuperación completa sin necesidad de intervenciones farmacológicas, algunos síntomas severos como la neuritis óptica con pérdida importante de la agudeza visual, el síndrome de tronco encefálico, el compromiso motor, ataxia y vértigo incapacitantes son susceptibles de manejo con altas dosis de corticoesteroides. El esquema que se utiliza con mayor frecuencia es metilprednisolona 1g cada día, por un periodo de tres a cinco días 51.
En el caso de la neuritis óptica, se sabe que los esteroides intravenosos disminuyen el tiempo de recuperación de la agudeza visual, pero no tienen ningún impacto sobre el prónostico visual a largo plazo ni sobre el riesgo de convertir a EM 52. Dentro de los efectos adversos de la terapia con esteroides se encuentran el aumento transitorio de la resorción ósea, hiperglicemia, hipertensión, dispepsia, manifestaciones psiquiátricas (depresión, ansiedad, psicosis), aumento de peso, insomnio, necrosis avascular del hombro, cadera y cataratas 53,54.
Tratamiento modificador de la enfermedad
Aunque ninguno de los tratamientos disponibles es totalmente efectivo en detener la actividad o la progresión de la enfermedad, existe evidencia disponible del impacto favorable del tratamiento después de un primer evento clínico.
El objetivo de la intervención terapéutica en los pacientes con SCA es evitar la actividad clínica futura, la acumulación de nuevas lesiones en la RM y la progresión de discapacidad a largo plazo. Varios estudios realizados demuestran que el inicio de tratamiento modificador de la enfermedad (TME) en pacientes con SCA de alto riesgo (RM cerebral con lesiones típicas de EM y BOC patrón tipo II o paciente que cumplen criterios de EM por RM) retrasa la conversión a EMCD. La decisión de iniciar o no TME en pacientes con SCA de alto riesgo debe individualizarse según las características y la presentación clínica, apoyados en los criterios diagnósticos y riesgo de conversión a EM 55.
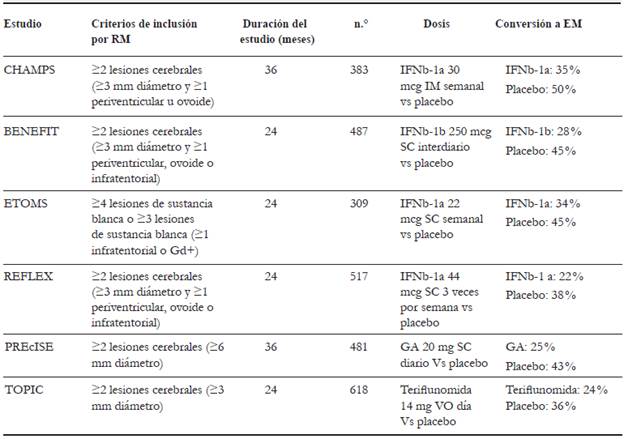
El primer estudio que demostró una disminución en la probabilidad acumulada de desarrollar EMCD con el uso de TME fue el estudio CHAMPS (Controlled High-risk subjects Avonex Multiple sclerosis Prevention Study), en el cual el uso de inferferon beta 1a intramuscular redujo esta probabilidad un 35 %%, en comparación con un 50 %% en el grupo placebo (p = 0,002) 38. En el estudio BENEFIT (Benefit in Newly Emerging MS for Initial Treatment Trial) se encontró una reducción del riesgo absoluto del 17 %% de convertir a EM clínicamente definida en brazo de pacientes tratados con interferon vs. el brazo placebo, con un efecto discreto en la discapacidad a cinco años 56.
Por otro lado, en el estudio PreCISe (Early GA Treatment in Delaying Conversion to CDMS in subjects Presenting with a Clinically Isolated Syndrome) el tiempo hasta la conversión a EMCD se redujo en un 25 %% en pacientes a los que se administró acetato de glatiramer, comparado con un 43 %% en el grupo placebo (p = 0,0005) 57.
El estudio de seguimiento ETOMS (The Early Treatment of Multiple Sclerosis) evidenció que el uso de IFN B1-a subcutáneo semanalmente redujo la conversión a EM a un 34 %%, en contraste con un 45 %% de conversión en el grupo control a dos años de seguimiento, tiempo en el que además se demostró una descenso en la tasa de atrofia cerebral respecto al grupo placebo 51.
Recientemente, varios estudios de extensión de las cohortes BENEFIT y CHAMPS mencionan el posible beneficio del inicio temprano de TME. En el ensayo clínico CHAMPS, 203 de los 383 pacientes fueron seguidos a cinco y diez años; el 36 %% de los pacientes en el brazo de tratamiento tuvieron una probabilidad acumulada de desarrollo a EM clínicamente definida, contra 49 %% de probabilidad en el grupo control a cinco años de seguimiento (p = 0,03), así como las nuevas lesiones en T2 fueron menos que en el grupo placebo 58. Con este estudio se logró concluir que retrasar el inicio temprano de TME por encima de tres años desde la manifestación clínica inicial conduciría a una conversión a EM de manera precoz; así mismo, no se demostró impacto estadísticamente significativo en la carga lesional ni en acumulación de discapacidad. Por otro lado, el estudio BENEFIT demostró una reducción en la discapacidad a tres años de tratamiento con interferon beta 1b instaurado de manera temprana, comparado con pacientes que iniciaron tratamiento de forma tardía. La administración precoz del interferon beta 1b interdiario por vía subcutánea redujo en un 37 %% el riesgo de conversión a EM (p = 0,003) 37.
El ensayo clínico TOPIC (Oral teriflunomide for patients with a first clinical episode suggestive of multiple sclerosis) concluyó que el inicio temprano de terifluno-mida 14 mg/día reduce en un 42,6 %% el riesgo de conversión a EMCD, en comparación con el grupo de control (p = 0,0087) (tabla 3) 59.
TABLA 3 TRATAMIENTO: BENEFIT, THE BETAFERON/BETASERON IN NEWLY EMERGING MS FOR INITIAL TREATMENT STUDY; CHAMPS, THE CONTROLLED HIGH-RISK SUBJECTS AVONEX MULTIPLE SCLEROSIS PREVENTION STUDY; ETOMS, THE EARLY TREATMENT OF MULTIPLE SCLEROSIS STUDY; GD+, LESIONES QUE REALZAN AL MEDIO DE CONTRASTE; IFN, INTERFERON; IMM, INTRAMUSCULAR; RM, RESONANCIA MAGNÉTICA; PRECISE, THE EARLY GLATIRAMER ACETATE TREATMENT IN DELAYING CONVERSION TO CLINICALLY DEFINITE MULTIPLE SCLEROSIS IN SUBJECTS PRESENTING WITH A CLINICALLY ISOLATED SYNDROME; REFLEX, THE REBIF FLEXIBLE DOSING IN EARLY MS STUDY; SC, SUBCUTÁNEO; TOPIC, THE ORAL TERIFLUNOMIDE FOR PATIENTS WITH A FIRST CLINICAL EPISODE SUGGESTIVE OF MULTIPLE SCLEROSIS STUDY
Muchos expertos, basados en los resultados de estos ensayos clínicos y su extensión de seguimiento, recomiendan el inicio de TME desde el SCA en pacientes con alto riesgo de conversión a EM; sin embargo, no todos los pacientes con SCA desarrollarán una EM, por lo tanto, es adecuado realizar el seguimiento clínico y radiológico en aquellos con buen pronóstico.
CONCLUSIONES
El síndrome clínico aislado corresponde al primer episodio clínico sugestivo de esclerosis múltiple remitente-recurrente, de al menos 24 horas de duración, que se presenta sin evidencia de procesos infecciosos, inflamatorios, metabólicos o encefalopatía asociados. Las manifestaciones clínicas típicas del SCA corresponden a la neuritis óptica unilateral, síndrome hemisférico, mielitis incompleta y síndrome de tronco encefálico. Existen factores predictores de riesgo para conversión a EM; los factores clínicos y demográficos son de bajo impacto, pero la RM y las BOC son herramientas de mayor valor predictivo en relación con el riesgo de conversión a EM. La decisión sobre iniciar o no agentes modificadores de la enfermedad en pacientes con síndrome clínico aislado debe ser individualizada teniendo en cuenta los factores predictores clínicos y paraclínicos disponibles en la actualidad.

