











 English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
Los errores innatos del metabolismo (EIM) corresponden a un grupo de enfermedades de origen genético, con gran heterogeneidad clínica, que causan alteraciones bioquímicas en una vía metabólica específica. La crisis convulsivas aisladas y la epilepsia se presentan en el 40-60 % de los pacientes. El 25 % de los pacientes convulsionan en el periodo neonatal y más del 90 % antes del final de la pubertad 1-8.La incidencia de los EIM es rara de manera individual, pero al día de hoy se han identificado aproximadamente 700 tipos. En conjunto su una incidencia es de 1:500 a 1:1000 recién nacidos 9-13. Su diagnóstico en países con limitación de recursos es laborioso, por lo que el propósito de este trabajo radica en considerar el electroencefalograma (EEG) como un elemento de ayuda en la aproximación diagnóstica.
El tratamiento se apoya en el empleo de fármacos anticonvulsivos, cofactores o una dieta especial para tratar y controlar sus manifestaciones 14-19. A continuación se describen las características electroclínicas en niños con EIM y epilepsia en una muestra obtenida del servicio de consulta externa y hospitalización del Hospital Universitario San Vicente Fundación (HUSVF) y el Hospital Pablo Tobón Uribe (HPTU) en Medellín, Colombia.
METODOLOGÍA
Estudio descriptivo de series de caso retrospectivo.
Población de estudio
Se incluyeron todos los registros de pacientes con diagnóstico de EIM en el periodo 2008-2017.
Criterios de inclusión
Edad menor de 18 años, criterio de epilepsia según la definición actual de la Liga Internacional contra la Epilepsia (ILAE), diagnóstico de enfermedad metabólica probable o confirmado según criterios clínicos, imagenológicos y de laboratorio, disponibilidad de la historia clínica y del trazado de EEG de superficie.
Aspectos éticos
El proyecto fue revisado y aprobado por los comités de ética del Instituto de Investigaciones Médicas de la Universidad de Antioquia, HUSVF y HPTU.
RESULTADOS
Se revisaron en total 211 historias, de las cuales 20 cumplieron los criterios de inclusión. En total fueron 13 pacientes masculinos y 7 femeninas, la edad de los pacientes se situó en un rango de 7 días a 15 años, con una mediana de 2 años. El 65 % procedía de áreas urbanas. Las variables clínicas analizadas se presentan en la tabla 1.
Tabla 1 Características clínicas en niños con EIM, HUSVF, HPTU, 2008 al 2017. Medellín, Colombia
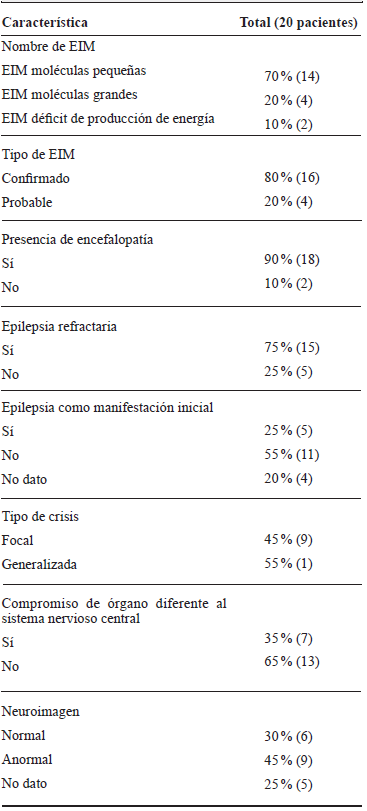
Fuente de los autores.
En cuanto a la clasificación clínico-molecular de los EIM, 70 % de los pacientes tenían enfermedad de molécula pequeña, 20 % de molécula grande y 10 % déficit de producción de energía. De los pacientes que tenían EIM de moléculas pequeñas, nueve tenían hiperglicinemia no cetósica (HGNC), dos enfermedad de la orina con olor a jarabe de arce, uno homocisteinuria, uno aciduriaglutárica, uno déficit de creatina y uno déficit de ornitintranscarbamilasa. De los EIM de moléculas grandes se identificó uno con adrenoleucodistrofia ligada al X, uno con enfermedad de Gaucher tipo neuropático y uno con déficit congénito de la glicosilación. De los EIM por deficiencia de producción de energía se encontró uno con encefalopatía mitocondrial con acidosis láctica y apoplejía (MELAS) y uno con enfermedad de Leigh. El 80 % de los pacientes tenía el diagnóstico confirmado genéticamente.
La mayoría de los pacientes tenía encefalopatía (90 %), el 55 % tenía diagnóstico de epilepsia, y de estos el 75 % tenía refractariedad a los medicamentos anticonvulsivos. El 45 % de los pacientes con EIM tenían anormalidades en la resonancia cerebral, siendo las más frecuentes: accidente cerebrovascular, edema cortical, polimicrogiria, aumento del espacio subaracnoideo temporosilviano y alteración de la señal ganglio basal y de la sustancia blanca.
En cuanto a las características EEG, se encontró que la mayoría tenía un ritmo de fondo anormal (90 %), 55 % tenían grafoelementos de sueño, pero el 81 % de ellos con mala estructuración; ninguno presentó fotosensibilidad. La actividad epileptiforme más común en EIM de moléculas pequeñas fue multifocal, mientras en EIM por déficit de producción de energía fue focal; no se encontró un patrón característico en EIM de moléculas de tamaño grande. Las variables EEG se especifican en las tablas 2 y 3.
Tabla 2 Características EEG en niños con EIM, HUSVF, HPTU, 2008 al 2017. Medellín, Colombia
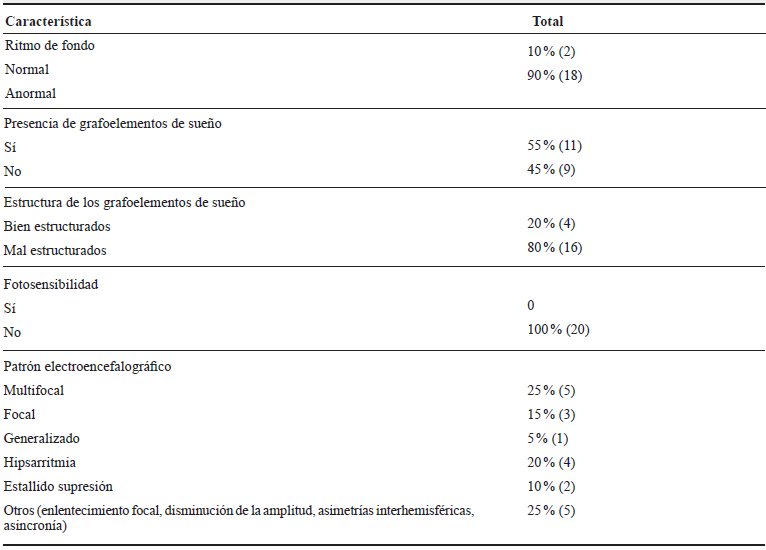
Fuente de los autores.
Tabla 3 Distribución del patrón EEG según tipo de EIM. HUSVF, HPTU, 2008 al 2017. Medellín, Colombia
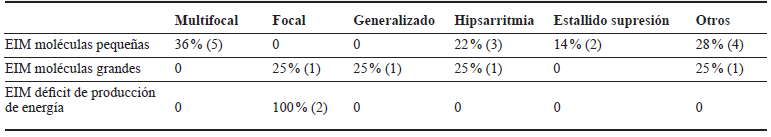
Fuente de los autores.
El 64 % de los pacientes con EIM de moléculas pequeñas tenía como diagnóstico hiperglicinemia no cetósica (HGNC), y en estos el patrón EEG más común entre 0 y 30 días era estallido supresión. De 30 días a 2 años predominó la hipsarritmia y en mayores de 2 años fue multifocal.
DISCUSIÓN
Los EIM son enfermedades con importancia creciente debido a que los pacientes son reconocidos más temprano, tienen mayor sobrevida y consultan a los servicios de urgencia por las mismas enfermedades prevalentes que afectan a su grupo de pares. Así mismo, los estudios de laboratorio están más disponibles 20,21. Todas estas razones han llevado a que el médico se concientice de su existencia y cómo intervenirlas apropiadamente.
Esta serie de casos estuvo compuesta principalmente por afectados por EIM de moléculas pequeñas, la mayoría de las cuales correspondía a HGNC. La región de procedencia de los pacientes de la muestra tiene quizá la mayor prevalencia mundial e influyó en los resultados presentados 22-24.
El motivo inicial de consulta fue retraso del neurodesarrollo, mientras que solo el 25 % de los pacientes consultó por convulsiones. Sin embargo, casi todos los pacientes evolucionaron a epilepsia refractaria 25,26.
En Colombia y Suramérica, el EEG es una herramienta con excelente relación costo-efecto, ayuda a orientar los estudios metabólicos iniciales y, en algunos casos, a sospechar si la etiología es genética, como en el síndrome de Angelman, Prader Willi, síndrome de Rett y mutación del SCN1 27-33.
La última propuesta de terminología de la ILAE incluye el término encefalopatía epiléptica o encefalopatía del desarrollo. El primero hace referencia al retraso y la regresión del desarrollo ocasionados directamente por la epilepsia. Los pacientes reportados en este estudio se agrupan preferencialmente en encefalopatía del desarrollo, lo que da a entender que hay un proceso subyacente a la epilepsia que interfiere en la adquisición de los hitos del desarrollo 34.
La mayoría de los pacientes reportados tenía crisis convulsivas motoras generalizadas y un grado variable de encefalopatía. Desde el punto de vista del EEG, la anormalidad electrográfica más común fue multifocal. Aunque el 55 % de los pacientes tenía grafoelementos hípnicos, en el 80 % de los casos estaban mal estructurados, asincrónicos, asimétricos, ausentes y entremezclados con actividad epileptiforme y disormia 35,36.
Con respecto a los afectados con HGNC, encontramos una ontogenia EEG particular del nacimiento a la niñez: en el periodo neonatal se presenta preferencialmente patrón estallido supresión, del mes a dos años hipsarritmia y luego de los dos años actividad epiléptica multifocal. En los pacientes con epilepsia y estos patrones EEG relacionados con la edad debería sospecharse HGNC 37,38.
En este estudio se reportaron cuatro casos no confirmados genéticamente, que se incluyeron para el análisis final porque tenían los elementos clínicos, bioquímicos y radiológicos compatibles con enfermedades mitocondriales (MELAS y leigh), moléculas grandes (Gaucher) y moléculas pequeñas (aciduria glutárica). En Colombia se necesitan estudios que diluciden las características genéticas patogénicas subyacentes a los EIM.
Nuestros resultados sugieren que el EEG es una herramienta que siempre debe considerarse en los EIM con compromiso neurológico 39,40.
Este estudio contó con todos los trazados EEG y permitió analizarlos con independencia de la interpretación inicial. Las limitaciones de este estudio consistieron en el tamaño de la muestra y que la recolección de los datos se hizo en forma retrospectiva.
CONCLUSIONES
El diagnóstico de los EIM está en ascenso por tener más accesibilidad a los métodos de pesquisa y sensibilización por parte de los profesionales de la salud. El perfil clínico molecular de este grupo de pacientes con EIM y epilepsia fue en su mayoría de molécula pequeña, sexo masculino, con retraso del desarrollo inicialmente y grados variables de encefalopatía del desarrollo. En cuanto a las variables EEG, la mayoría tenía un ritmo de fondo anormal, con grafoelementos de sueño mal estructurados y el patrón EEG dependiente del tipo de EIM. El EEG es una herramienta costoefectiva para orientar los estudios iniciales en los pacientes con sospecha de EIM.

