











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
La epilepsia es una de las enfermedades neurológicas crónicas más comunes, afectando a más de 50 millones de personas en el mundo. Cada año, 80 por cada 100.000 personas desarrollan epilepsia de novo, siendo más común su inicio en la infancia 1. La mayoría de los pacientes diagnosticados reciben tratamiento con antiepilépticos, pero una gran proporción continúan teniendo crisis a pesar de un manejo adecuado, lo que establece el concepto de epilepsia resistente o refractaria, la cual puede deteriorar la calidad de vida del paciente, llevando a la búsqueda de alternativas terapéuticas como la cirugía de epilepsia, dieta cetogénica o estimulación del nervio vago 2. De acuerdo a los criterios de la Liga Internacional contra la Epilepsia (ILAE), se define epilepsia refractaria como aquella en la cual se ha producido el fracaso a 2 ensayos de fármacos antiepilépticos (FAE), en monoterapia o en combinación, tolerados, apropiadamente elegidos y empleados de forma adecuada, para conseguir la ausencia mantenida de crisis 3,4.
En años recientes, el progreso en el entendimiento de las bases genéticas de las epilepsias generalizadas idiopáticas y refractarias ha probado ser un reto, debido a los complejos patrones de herencia y la heterogeneidad genética y clínica. El polimorfismo genético ofrece una ventaja en la compresión etiológica de estos tipos de epilepsia pues cumplen un papel patogénico. Muchos de éstos codifican importantes estructuras de los canales de sodio en el sistema nervioso central, mientras que otros codifican para importantes rutas metabólicas 5.
Uno de los genes más estudiados en la etiología de epilepsias idiopáticas y algunos síndromes epilépticos es el SCN1A (Sodium Channel, Neuronal Type I, Alpha Subunit), que codifica la subunidad alfa de los canales neuronales de sodio dependientes de voltaje (NaVl.1), se expresa predominantemente en interneuronas gabaérgicas y hace parte de un agrupamiento de genes ubicados en el cromosoma 2q24.3, que incluye también los genes SCN2A y SCN3A 6. La mayoría de las mutaciones asociadas con epilepsia que pueden ser detectadas alteran el extremo C terminal del canal, llevando a disfunción neuronal e hiperexcitabilidad en conexiones corticales y cerebelosas 7.
Las mutaciones del gen SCN1A típicamente son mis-sense (mutación de una sola base que genera la producción de una forma alterada de la proteína) o frameshift (forma más frecuente), además 80 a 90% de las mutaciones son de novo, y en el caso de las heredadas son de tipo autosómico dominante pero con penetrancia incompleta y expresividad variable, con mosaicismo en hasta un 7%, lo que permite explicar, entre otros factores, la expresión parcial o existencia de portadores asintomáticos 7,8. La prevalencia global de estas mutaciones es desconocida 8.
Los trastornos epilépticos asociados al gen SCN1A incluyen entre otros: epilepsia generalizada con convulsiones febriles plus, epilepsias mioclónicas atónicas, síndrome de West, síndrome de Landau-Klefner y especialmente epilepsia mioclónica severa en la infancia (síndrome de Dravet); esta última, descrita desde hace más de 40 años 9, es una condición infrecuente, con una tasa estimada de 1 caso por cada 40.000 niños, y en la cual se demuestran mutaciones del gen SCNA1 en aproximadamente el 80% de los pacientes 8. Se considera una encefalopatía epiléptica caracterizada por aparición temprana de crisis mioclónicas, crisis tónico-clónicas generalizadas (TCG) y ausencias atípicas, así como estatus epiléptico refractario desde el primer año de edad, siendo en general de pobre pronóstico cognitivo y muy difícil control, considerándose como el fenotipo más grave de dicho conjunto, sin embargo, existe aún incertidumbre en cuanto a su evolución a largo plazo 8,10.
A continuación, se presenta el caso clínico de una paciente de 10 años con epilepsia refractaria y mutación en el gen SCN1A.
Presentación de caso
Paciente femenina de 10 años al momento de la elaboración del artículo, fruto de la primera gestación de bajo riesgo, controlada, sin complicaciones perinatales y sin antecedentes maternos de importancia. Nace a término por cesárea programada, presenta circular a cuello no apretada, con adaptación neonatal espontánea, sin complicaciones.
La paciente presentó inicialmente un neurodesarrollo adecuado en sus primeros meses de vida sin otros antecedentes patológicos, alérgicos, ni quirúrgicos; vacunación completa sin inconvenientes. Sin embargo, a la edad de 6 meses, posterior a la administración de una dosis de vacuna DPT presentó una primera convulsión febril TCG de 10 minutos de duración incluyendo el posictal, sin evidencia de complicación ni neuroinfección. En los siguientes meses presentó regularmente convulsiones febriles, incluso hasta 1 día antes del pico febril, asociados tanto a vacunaciones como a procesos infecciosos, pero sin convulsiones complejas ni deterioro progresivo del estado neurológico.
Después del año la paciente empezó a presentar episodios de inicio súbito caracterizados por desconexión del medio, fijación de la mirada, y parpadeo, en ocasiones palidez central, todo esto seguido de extensión tónica y luego movimientos tónico-clónicos de extremidades. Estos episodios frecuentemente progresaron a estatus epiléptico de hasta una hora y media, sin control tras el inicio de topiramato, lamotrigina ni oxcarbazepina, acumulando aproximadamente 22 crisis focales y generalizadas con o sin picos febriles, con progresión a estatus con múltiples hospitalizaciones en unidad de cuidado intensivo, en los primeros a 2 años de vida. Adicionalmente, asociado a las crisis, comenzó a presentar retraso en el neurodesarrollo predominantemente en lenguaje expresivo e integración sensorial. Debido a la poca respuesta con diferentes regímenes terapéuticos y los cuadros infecciosos recurrentes que incluso comprometieron sistema gastrointestinal causando una necrosis isquémica de intestino delgado de origen infeccioso, se suspendieron estos y se inició manejo con inmunoglobulina G cada 3 semanas bajo impresión de una inmunodeficiencia primaria (buscando reducir los episodios febriles) por 5 meses y clonazepam en caso de crisis.
La paciente continuó presentando cuadros infecciosos a repetición, requiriendo múltiples hospitalizaciones asociados a crisis epilépticas de presentación variable, pero comúnmente con posterior generalización y asociación a síntomas neurovegetativos. En ocasiones se presentaban con versión cefálica izquierda, supraversión de la mirada, midriasis, palidez y luego movimientos tónicos generalizados, o en ocasiones clonías alternantes de extremidades superiores, con parpadeo bilateral y nistagmo. Algunos de estos episodios se precedieron de gritos y fueron más acentuados en las horas de la mañana; en otras ocasiones, se presentaban con arresto motor y mirada fija, otras veces con clonías de hemicara, alternantes izquierda o derecha y en general con patrones variables. La exacerbación de las crisis requirió ajuste de esquemas terapéuticos por lo que se reinició topiramato y se adicionaron ácido valproico y clobazam.
Inicialmente, se realizaron múltiples electroencefalogramas convencionales con resultados normales. Un video-EEG de 12 horas reportó actividad irritativa en la región central, con diseminación a ambas regiones fronto-centrales, siendo este el hallazgo más llamativo y consistente en estudios posteriores (figura 1).
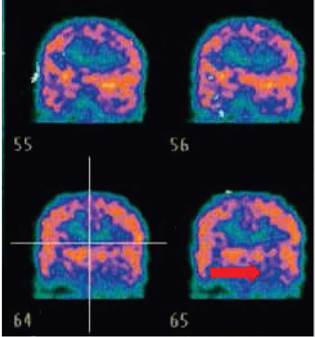
Figura 1 Tomografía por emisión de positrones. Tomografía por emisión de positrones (PET-CT). Indicador señala alteraciones en el metabolismo en región temporal izquierda. Tomado de soportes de historia clínica provistos por la acudiente de la paciente.
Durante el estudio de la epilepsia se solicitaron múltiples paraclínicos que incluyeron, además de resonancia magnética cerebral con espectroscopia cuyo resultado fue normal, una nueva telemetría de 96 horas que mostró 3 crisis focales (figura 2) con actividad epileptiforme frontal bilateral con predominio basal derecho y generalización secundaria. Se realizó PET-CT que evidenció moderada a severa disminución del metabolismo en región temporal izquierda. Adicionalmente se hizo estudio completo de inmunodeficiencias y enfermedades metabólicas que cuáles fueron negativos (figura 3).
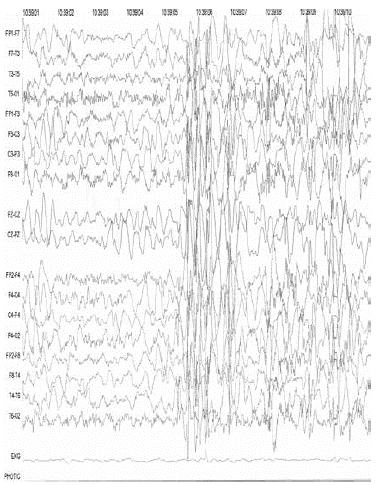
Figura 2 Eventos ictales en Video monitoreo EEG. Parte del trazado de VideoEEG de 12 horas que evidencia actividad irritativa en la región central con diseminación bilateral. Tomado de soportes de historia clínica provistos por la acudiente de la paciente.
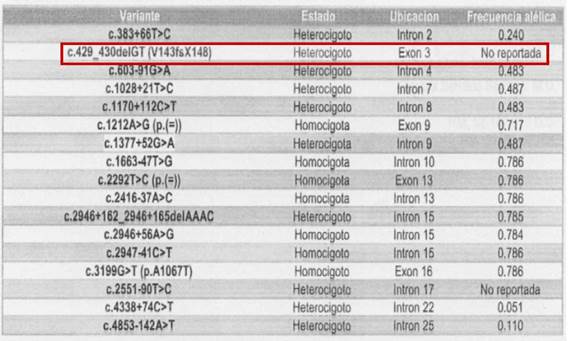
Figura 3 Análisis por microdeleciones evidenciando la mutación. Tomado de soportes de historia clínica provistos por la acudiente de la paciente.
Posteriormente presentó alteraciones de la conducta, se tornaba irritable, con lenguaje ecolálico en asociación a persistencia de estatus convulsivos de hasta 3 horas y media de duración.
Debido a la refractariedad, severidad y recurrencia tanto de crisis como de infecciones, se sospechó una posible canalopatía. Fue valorada por genética clínica y se le realizó estudio de mutaciones del gen SCN1A o 1B para canales de sodio o GABA mediante secuenciación de la región codificante del gen SCN1A, y se encontró que la paciente era portadora de la mutación c.429_430delGT (V143fsX148), que consiste en pérdida de dos bases (GT) (figura 4), en las posiciones 429 y 430, lo cual genera un desplazamiento en el marco de lectura y da origen a un codón de parada prematuro en la posición 148 de la proteína. Esta mutación, ubicada en el exón 3 ha sido previamente descrita y ya ha sido reportada su asociación con el síndrome de Dravet (SD).
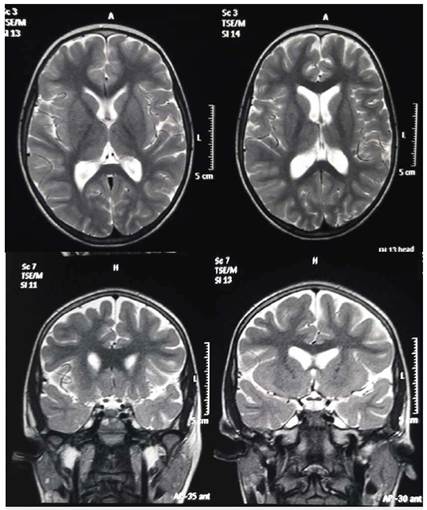
Figura 4 Resonancia Magnética Nuclear de cerebro. Tomado de soportes de historia clínica provistos por la acudiente de la paciente.
A la edad de 5 años, se continuó esquema de ácido valproico, topiramato e IgG humana (nuevamente bajo impresión diagnóstica de una inmunodeficiencia primaria que ocasionaba cuadros febriles recurrentes), presentando mejoría progresiva de las crisis, adicionalmente, mejoría notoria en el lenguaje y disminución de los episodios infecciosos. Se continuó con desmonte escalonado de medicamentos con crisis cada vez más espaciadas y especialmente asociadas a episodios febriles.
Se inició proceso interdisciplinario de rehabilitación a cargo de neuropsicología y terapia ocupacional, así como implementación de adecuaciones curriculares. Actualmente se encuentra en segundo grado de básica primaria, en proceso de desmonte de ácido valproico, con parámetros de desarrollo psicomotor y neurológico concordantes para su edad permitiéndole una mejoría notoria en su calidad de vida.
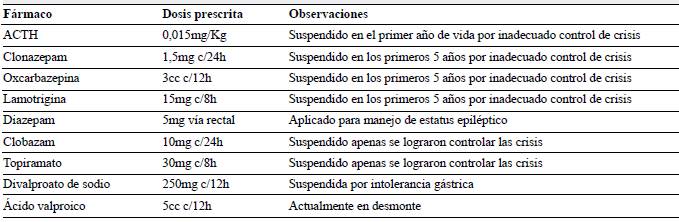
En la tabla 1 se puede ver el resumen de los antiepilépticos prescritos a la paciente.
DISCUSIÓN
La epilepsia de origen genético corresponde hasta a 20% de las epilepsias. Un estudio retrospectivo realizado en Medellín, Colombia describió que 53 de 5.357 pacientes evaluados en un centro de neuropediatría (1% de la población) presentan epilepsia genética generalizada, 21% de los casos de fenotipo variable, como la paciente descrita 11. Las manifestaciones clínicas derivadas de mutaciones del gen SCN1A, y en particular el síndrome de Dravet en sus presentaciones típicas y atípicas, se asocian a un pobre pronóstico debido a las características de las crisis, al deterioro cognitivo, así como a una alta mortalidad 8,10-14.
La mayoría de pacientes presentan un retraso del desarrollo que tiene inicio al momento de la primera convulsión, es descrito más como un freno en la ganancia de hitos del neurodesarrollo que como una regresión progresiva, puesto que el compromiso, inicialmente precipitado, suele remitir luego de los primeros 5 años de vida y estabilizarse al finalizar la primera década, pero en general, el compromiso ya establecido nunca se recupera por completo 10-12.
El caso actual presenta una historia y evolución en concordancia con lo que se ha establecido para una presentación típica de un síndrome de Dravet 9-11, el tiempo de inicio, los desencadenantes, tipos y características de las crisis, la presencia de estatus epilépticos refractarios y recurrentes, la ausencia de anomalías electroencefalográficas iniciales, la aparición de un desfase del desarrollo y la detección de una mutación puntual del SCNA1, entre otros, establecen dicha impresión. Llama la atención que luego de los primeros 5 años, presenta una significativa evolución a la mejoría dado por un adecuado control de crisis, con desmonte de medicamentos y de igual forma, presentando estabilización del compromiso neuropsicológico, permitiendo que, con un proceso interdisciplinario de rehabilitación la paciente comience a presentar ganancia de habilidades y reducción significativa del desfase del neurodesarrollo, así como una mejoría significativa en su calidad de vida.
Desde las primeras descripciones del síndrome de Dravet, múltiples estudios observacionales han identificado casos con pronósticos más favorables e incluso de remisión completa de crisis, así como de un notable menor compromiso intelectual, que aunque infrecuentes, han aumentado en su proporción, especialmente en los de más reciente diagnóstico 11-15.
Un estudio de cohorte realizado por Takayama et al 10, en el que se realizó un seguimiento durante 2 a 3 décadas a 64 pacientes separados en 2 grupos de acuerdo a sus características clínicas y paraclínicas, identificó algunas diferencias estadísticamente significativas en los pacientes de mejor pronóstico. La presentación clínica, características del EEG y de las mutaciones fueron las variables de mayor relevancia. Solo 1 de los pacientes con presentación típica presentó remisión de crisis mientras que en el grupo de presentación atípica lo lograron 4 pacientes (2,3% y 20% respectivamente p = 0,03). En lo que respecta al EEG, la ausencia de descargas epileptiformes y la aparición de ritmos alfa occipitales tuvo asociación significativa con remisión de crisis (valores p = 0,0004 y 0,001 respectivamente) 10. Este último hallazgo también ha sido corroborado en revisiones como la de Akiyama et al 16.
En cuanto al desenlace cognitivo descrito en el mismo trabajo, en línea con lo ya establecido en la literatura, todos los pacientes presentaron déficit cognitivo, la mayoría moderado a grave y solo 3 con déficit leve. La mayor frecuencia de crisis TCG y las presentaciones típicas se asociaron de manera significativa con mayor compromiso cognitivo (valores p = 0,0019 y 0,0283 respectivamente). En concordancia, otros autores como Kim et al 15 y Genton et al 14, han evidenciado que con frecuencia, en las presentaciones atípicas el deterioro cognitivo es más tardío y de menor severidad 15. A su vez, la aparición de ritmos alfa occipitales se asoció también a un menor grado de déficit cognitivo (p = 0,0085) 16.
Debe mencionarse que, a pesar de lo descrito previamente, algunos estudios no evidencian asociaciones significativas entre frecuencia y edad de presentación de crisis con un mayor compromiso cognitivo 17,18. Por lo tanto, se ha sugerido que el compromiso cognitivo y neuropsicológico puede tener relación y origen en factores diferentes a las crisis epilépticas 8,11,12.
Se ha establecido que la mutación del SCN1A conlleva a un funcionamiento anormal de la actividad eléctrica en el sistema nervioso central que así como epilepsia, por sí mismo puede dar lugar a compromiso cerebeloso (frecuentemente descrito en pacientes con SD) 8, alteraciones del neurodesarrollo e incluso se ha postulado su papel en la etiología de trastornos psiquiátricos 19,20, esto último de la mano del concepto de canalopatías, entendidas como un conjunto heterogéneo de alteraciones con una expresión muy variable, con muchas características aún por definir 11,12,19.
La paciente del caso difiere de lo reportado en que su presentación es típica y aunque leves, presenta descargas epileptiformes en los EEG de control, por lo que no se esperaría una evolución tan favorable. Además, no presenta varios de los factores ya descritos que se asocian a mejor pronóstico. Podría considerarse, por lo tanto, que la evolución positiva de la paciente en los componentes cognitivo y del neurodesarrollo puede deberse a que estos no dependen únicamente de la gravedad de las convulsiones pues otras variables ejercen influencia, como el tipo de mutación (frameshift vs missense) 8,12, la porción afectada de la proteína (compromiso de la región formadora de poro), presencia de mutaciones concomitantes de genes modificadores como el CACNB48,10, factores epigenéticos y ambientales, diferentes modelos de herencia, así como la presencia de múltiples fármacos antiepilépticos (algunos como lamotrigina o carbamazepina pueden exacerbar las crisis) y limitaciones al acceso a terapias de rehabilitación y estimulación 21. Todo en línea con la heterogeneidad e incertidumbre en la evolución de las canalopatías.
Cabe mencionar también que el reconocimiento temprano de la enfermedad, así como el manejo más agresivo de las crisis (terapia combinada con topiramato y clobazam), pueden afirmarse como una de las posibles causas de la mejoría en la paciente, ya que se ha establecido previamente en series longitudinales que en pacientes de reciente diagnóstico hay mejor control de crisis y menor compromiso cognitivo en quienes se inicia terapia más tempranamente 13,15,22.
Casos como el actual apoyan la idea de que aún son necesarios más estudios que permitan ampliar el entendimiento de patologías tan diversas como las surgidas a partir de estas mutaciones, puesto que los determinantes de su pronóstico son muy diversos proporcionan gran incertidumbre. De igual forma, recalca la importancia de considerar un diagnóstico genético precoz en pacientes con epilepsias refractarias de inicio temprano, incluso en aquellos con presentaciones atípicas, pues su curso y pronóstico son variables y podrían impactarse con una detección y tratamiento interdisciplinario tempranos 8,12,19,21.
CONCLUSIONES
El síndrome de Dravet corresponde a un subconjunto de las epilepsias refractarias, asociadas a mutaciones del gen SCNA1. Su pronóstico clínico y cognitivo es desfavorable, sin embargo, hace parte del ampliamente diverso grupo de las canalopatías sobre las cuales hay aún grandes incertidumbres. La gravedad de las crisis y del compromiso neuropsicológico pueden estar determinados por múltiples factores diferentes a la refractariedad de las crisis. Este caso, y la evidencia actual, apoyan la relevancia de la sospecha y determinación de un diagnóstico genético en pacientes con epilepsias refractarias tempranas, incluso en las presentaciones atípicas, puesto que permite aclarar la etiología del cuadro y definir un tratamiento oportuno para impactar significativamente en el pronóstico.

