











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
PermalinkINTRODUCCIÓN
Las lipofuscinosis ceroides neuronales (CLN) son un grupo de enfermedades neurodegenerativas caracterizadas por acúmulo intracelular de material fluorescente, en los liso-somas de células neuronales del cerebro y retina 1. Hasta la fecha se han descrito 8 diferentes tipos de la enfermedad causados por la mutación de 13 genes, aunque existe reporte en la literatura de hasta 14 mutaciones (CLN1-CLN14). La Academia Americana de Neurología describe la nueva nomenclatura y esquema de clasificación para las CLN de acuerdo al defecto genético subyacente (Tabla 1) 2.
Tabla 1 Clasificación genética de la lipofuscinosis ceroidea neuronal.
| NOMBRE LOCUS | ESPECTRO FENOTIPICO | SIMBOLO GEN | LOCUS | NOMBRE PROTEINA |
|---|---|---|---|---|
| CLN 1 | I, LI, J, A | PPT1 | 1p34.2 | Proteina Palmitoil Tioesterasa 1 |
| CLN 2 | LI,J P | TPP1 | 11p15.4 | Tripeptidü Peptidasa 1 |
| CLN 3 | J, P | CLN3 | 16p11.2 | CLN3 |
| CLN 4 | A (Enfermedad de Parry) | DNAJCS | 20q13.33 | DnaJ homologo |
| CLN 5 | LI, J , P, A | CLN5 | 13q22.3 | CLN5 |
| CLN 6 | LI, P, A (Enfermedad de Kufs) | CLN6 | 15q23 | CLN6 |
| CLN 7 | LI, J | MFSDS | 4q2S.2 | Superfamilia facilitadora principal |
| CLN S | LI, P | CLNS | Sp23.3 | Cln8 |
| CLN 9 | Desconocido | N/A | Desconocido | Desconocido |
| CLN 10 | C, LI, J, A | CTSD | 11p15.5 | Catepsina D |
| CLN 11 | A | GRN | 17q21.31 | Granulinas |
| CLN 12 | J ATPasa 13A2 | ATP13A2 | 1p36.13 | Probable transporte de cationes |
| CLN 13 | A | CTSF | 11q13.2 | Catepsina F |
| CLN 14 | I | KCTD7 | 7q11.21 | BTB/POZ |
C: congénito; I: Infantil (6-24 meses); LI: Infantil tardío (2-3 años); J: Juvenil (5-7 años); A: adulto; P: Prolongado.
Adaptado de: Orsini A, Valetto A, Bertini V, et al. The best evidence for progressive myoclonic epilepsy: A pathway to precision therapy. Seizure. 2019;71:247-257.
Hasta el momento se desconoce la función del gen CLN6; sin embargo, codifica una proteína de 27KDa con 311 aminoácidos localizada en la membrana del retículo endoplásmico de la mayoría de los tejidos, especialmente cerebelo e hipotálamo; algunos estudios sugieren participación en la homeostasis lisosomal, al regular el transporte selectivo de proteínas y lípidos, la regulación de la acidificación celular, endocitosis y autofagia, elementos fundamentales en la función de este 2,3.
En la actualidad, se han descrito alrededor de 130 pacientes con mutaciones en el gen CLN6, en la cual se describen 3 variantes clínicas: De inicio infantil tardío o juvenil precoz, variante Kufs tipo A y Kufs tipo B 2. El subtipo de inicio infantil tardío o juvenil precoz se presenta con convulsiones, regresión del desarrollo, disartria, ataxia y pérdida de visión 2. En el subtipo Kufs tipo A, la enfermedad se inicia entre los 12 y los 51 años, con edad media de presentación de 28 años, el cuadro clínico se presenta con dificultades escolares, epilepsia mioclónica progresiva y menos frecuentemente crisis tónico clónicas generalizadas, deterioro conductual, ataxia y distonía, sin afección retiniana 2. El subtipo Kufs B cursa con deterioro cognitivo, demencia, síntomas motores cerebelosos o extrapiramidales, algunos casos con variantes patogénicas en el gen catepsina F (CTSF) 2.
Reporte de caso
Hombre de 33 años de origen colombiano, mestizo; a los 29 años presentó crisis tónico clónica generalizada de 3 minutos de duración. El electroencefalograma fue anormal, y la resonancia magnética (RM) mostró atrofia cerebral. En ese momento se atribuyó a evento perinatal, y se inició manejo con ácido valproico; estuvo libre de crisis por dos años. Presentó nuevo episodio ictal de similares características, coincidiendo con presencia de movimientos anormales tipo temblor; se cambió de medicación a levetiracetam con control de episodios convulsivos.
Es producto del primer embarazo, cursó con oligoamnios de etiología no establecida, trabajo de parto prolongado por 48 horas, parto espontáneo en cefálico, llanto y respiración espontánea, cianosis leve, pesó 3750 g, talla 52 cm, no patología neonatal. Sin antecedentes patológicos previos. Padres sanos, no consanguinidad y una hermana sana. Un primo y prima materna en segundo grado con epilepsia, desconoce el tipo de crisis; primo materno con hipoacusia neurosensorial (figura 1).
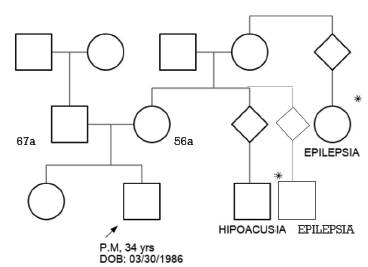
Fuente: Elaboración de los autores.
Figura 1 Pedigree 3 generaciones. Mostrando carácter recesivo de patología asociada a epilepsia.
Su neurodesarrollo estuvo acorde a su edad hasta los 14 años; ingresó al colegio a los 4 años con rendimiento promedio en escolaridad primaria y regular en secundaria, evidencian dificultad en la interacción social. Inició estudios universitarios sin concluirlos e inició y finalizó estudios técnicos, notan lentitud motora y del habla. Inicia vida laboral con dificultad en la adaptación, por falta de rendimiento y olvido en las actividades.
Al examen físico antropometría normal, signos vitales estables; buenas condiciones generales, buena actividad espontánea. Esfera mental inferior para su edad, conducta pueril, diestro, orientado en persona y tiempo, disartria, realiza analogías, fallas en abstracción, pensamiento concreto. Aumento de la pigmentación de la retina, rojo retiniano normal; reflejos músculo tendinosos exaltados en las 4 extremidades con reflejos pendulares rotulianos, respuesta plantar extensora bilateral, sensibilidad y fuerza normal, ligero aumento del tono muscular en miembros inferiores; dismetría y disdococinesia más del hemicuerpo izquierdo, temblor fino en ambas manos. Marcha sin aumento del polígono de sustentación, Romberg positivo, marcha en puntas con inestabilidad.
Valorado por neurología y genética, los estudios metabólicos fueron normales, cariotipo 46XY, metales pesados plomo, arsénico, cobre, y mercurio normales, función tiroidea normal. Nuevo estudio de RM muestra atrofia cerebral y cerebelar (figura 2), videotelemetría no registró actividad epileptiforme, y tomografía de coherencia óptica normal; campos visuales derecho e izquierdo obligan a descartar glaucoma.
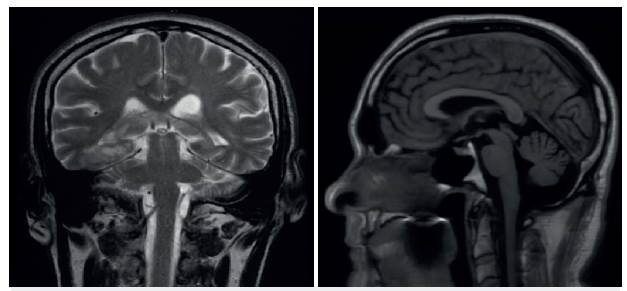
Fuente: Archivo de los autores.
Figura 2 Resonancia magnética cerebral simple. Secuencia coronal T2 y sagital T1. Evidenciándose atrofia cortical con discreta dilatación de ventrículos. Discreta atrofia de las folias cerebelosas.
Por cuadro de deterioro neurológico progresivo, epilepsia y neuroimágenes con atrofia cortical y cerebelar, se solicita secuenciación exómica con resultados de dos variantes heterocigotas para el gen CLN6, una patogénica y otra de significado incierto; con esto se realiza diagnóstico clínico y genético de lipofuscinosis ceroidea neuronal 6 (CLN6) variante clínica Kufs tipo A. El paciente ha continuado sus controles por neurología, presenta trastorno del comportamiento y es dependiente en algunas actividades básicas de la vida cotidiana.
DISCUSIÓN
Las lipofuscinosis ceroides neuronales (CLN) corresponden a un grupo de trastornos neurodegenerativos hereditarios monogénicos, ocurren principalmente en la primera década de la vida 4. Se han identificado catorce tipos de CLN, siendo la CLN3 la más común de estas entidades; cada uno de los diferentes tipos es causado por mutación en diferentes genes, los diferentes tipos de CLN son autosómicos recesivos, excepto la CLN4 de aparición en adultos 5. En este paciente fue identificado por estudio genético una variante patogénica en el gen CLN6, uno de los genes identificados como causante del subtipo infantil tardío 10.
Patológicamente, estos trastornos son neurodegenerativos y comparten un sello común de acumulación de material autofluorescente en los lisosomas, llamado ceroidelipofuscina, con apariencia ultraestructural típica bajo microscopía electrónica, al parecer no relacionada directamente con pérdida neuronal 6. La evolución tórpida y el componente de neurodegeneración se pudo evidenciar en este caso, por presencia de síntomas cerebelosos e involución cognitiva asociados a cambios anatómicos de atrofia cerebelosa y cerebral, evidenciados en las neuroimágenes.
Las CLN comparten presentación clínica similar caracterizada por convulsiones, pérdida visual disminución de las capacidades cognitivas y motoras debido a muerte neuronal progresiva 4. Estos trastornos muestran variación, especialmente en la edad de inicio, tasa de progresión de la enfermedad y primeros síntomas 4. En nuestro caso, el paciente inició con alteraciones de las capacidades cognitivas y convulsiones, no ha presentado deterioro visual; con reporte de estudio ocular que describe sospecha de glaucoma, asociación clínica no reportada en la literatura por lo que se presume puede ser un hallazgo incidental.
Además de la clasificación de acuerdo al defecto genético subyacente (Tabla 1) 2,7,8, se han descrito clasificaciones por correlación fenotipo-genotipo de acuerdo con la edad de inicio 9. La CLN6 puede iniciarse en la etapa infantil tardía, adolescencia o adultez (12-51años), conocida como variante o enfermedad de Kufs, en quienes la pérdida visual generalmente se encuentra ausente y presentan epilepsia mioclónica progresiva (tipo A) o demencia con deterioro motor (tipo B) generalmente alrededor de los 30 años 10. Descrita por primera vez en la India, en la península ibérica, en América Central y del Sur, presenta una distribución mundial con una prevalencia de 1:14.000 a 1:100.000 1. La mutación ocurre en el gen que codifica una proteína de membrana de 311 aminoácidos ubicada en el retículo endoplásmico, cuya función es desconocida; su ausencia afecta la degradación de la proteína índice aritosulfatasa A endocitosada 1,9. Estas características clínicas, fueron presentadas por nuestro paciente en el curso de su enfermedad; configurando un diagnóstico clínico de la variante CLN6 tipo Kufs A.
Las mutaciones recesivas en CLN6 12,13 y las mutaciones dominantes en DNAJC5 14,15 pueden causar Kufs tipo A; Kufs tipo B puede ser causado por mutaciones recesivas en CTSF 16,17. Se ha descrito que la enfermedad de Kufs tipo A identificada en el paciente, inicia sus síntomas a los 14-15 años con dificultades escolares y en la interacción social que progresan con la edad; posteriormente con presencia de convulsiones generalizadas sin presencia de mioclonías; con identificación exómica de dos variantes heterocigotas para el gen CLN6, una de ellas patogénica.
Sharp et al. en 2003 18, describieron una serie de 7 pacientes con CLN6 en 3 familias de Costa Rica, la mayoría homocigotos para E72X en el exón 4, con síntomas entre 5 y 7 años, ocurriendo la muerte entre los 14,5 y los 16,8 años; presentándose más tardíamente en comparación con familias reportadas en el mismo estudio de Portugal y Pakistán en donde se reportó mortalidad a los 10,5 años y 9 años respectivamente 18. En este estudio no se hace diferenciación entre el tipo de variante CLN6; llama la atención que nuestro paciente a la valoración con 33 años, supera estos rangos etáreos, mostrando lo heterogéneo en el fenotipo de las CLN.
Elleder et al. en 1997 19; informaron una serie de casos de CLN6 en República Checa que incluía a 27 niños de 23 familias, la edad de inicio estuvo entre los 18 meses y los 8 años. La alteración de la marcha, el habla, las convulsiones y el retraso del desarrollo fueron características tempranas. La enfermedad progresó rápidamente a la muerte entre los 18 y 32 años. El EEG mostró una respuesta a la fotoestimulación de manera temprana (2 a 4 años) 19.
Con el advenimiento de técnicas genéticas se pueden realizar diagnósticos específicos de CLN6 utilizando métodos mínimamente invasivos. El EEG puede ser útil en el diagnóstico temprano, el hallazgo de punta onda después de la fotoestimulación en niños con inicio de convulsiones debería hacer investigar la enfermedad CLN, especialmente los tipos CLN2, se ha visto el mismo patrón de punta en los pacientes con CLN6. En el caso descrito no se presentaron descargas asociadas a estimulación fótica, la RM mostró atrofia discreta cortical y de folias cerebelosas; el diagnóstico diferencial en pacientes pediátricos con crisis convulsivas o mioclonías, en los que se han descartado infecciones, neoplasias o malformaciones, conduce a la investigación de los diferentes tipos de epilepsias mioclónicas progresivas, dentro de las cuales se encuentran las CLN, que son el grupo más común de enfermedades neurodegenerativas de depósito lisosomal en la infancia 13.
Canafoglia et al. en 2015 13; presentaron 3 pacientes con CLN6, con inicio de síntomas en la adultez, estos pacientes debutaron con mioclonías y el EEG registrado a la edad promedio de 28,6 años mostró una organización de fondo preservada con presencia de bandas alfa en las frecuencias inferiores, pocas veces con actividad rápida en las derivaciones posteriores. Alteraciones del EEG no evidenciadas en este paciente 13. La RM no tiene buena sensibilidad ni especificidad para diagnóstico temprano, sin embargo, es útil para seguimiento de la progresión de la enfermedad 10. Presentamos un caso con inicio similar de convulsiones en la segunda década de la vida, con reporte de EEG normal y donde en el control de neuroimagen se pudo evidenciar el progreso de un proceso neurodegenerativo de base, confirmado posteriormente.
El panorama de estos trastornos ha cambiado en los últimos años con la primera terapia de reemplazo enzimático aprobada por la FDA en pacientes con CLN2, que mejora significativamente la calidad de vida y retrasa la progresión de la enfermedad 11,20. A la fecha no existe tratamiento curativo para CLN6, el tratamiento es sintomático enfocado al mantenimiento de una buena calidad de vida 21.
En casos de pacientes con dificultades escolares en adolescencia, con posterior deterioro cognitivo, dificultades en la interacción social; signos cerebelosos, seguidos de inicio de convulsiones mioclonicas o tónico clónicas generalizadas sin degeneración retiniana; se debe considerar la enfermedad de Kufs tipo A y la solicitud de estudios genéticos para mutaciones en el gen CLN 6.

