











 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mail Cited by SciELO
Cited by SciELO  Cited by Google
Cited by Google  Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google 
 Permalink
Permalink
Introducción
La enfermedad por anticuerpos contra la glicoproteína de la mielina de los oligodendrocitos (del inglés MOGAD) es un síndrome de reciente descripción en el espectro de las enfermedades inflamatorias desmielinizantes (EID) del sistema nervioso central (SNC). La glicoproteína de mielina de oligodendrocitos (MOG) se identificó como uno de los componentes de la mielina desde la década de 1980 1, y en los humanos se encuentra exclusivamente en el SNC. Su gen se localiza en el brazo corto del cromosoma 6, en relación con el complejo mayor de histocompatibilidad 2. Aunque su función es hasta ahora desconocida, se plantea que puede actuar como receptor del oligodendrocito 3,4 o ser un indicador de su maduración 5. Si bien los anticuerpos anti-MOG se conocen desde los años ochenta, no se ha logrado establecer su rol patogénico en la esclerosis múltiple (EM) o si influyen en su curso clínico, diagnóstico o seguimiento. Se ha demostrado que los anticuerpos anti-MOG están presentes en el suero de una minoría de pacientes con EM 6,7 y frecuentemente en sujetos sanos 8. En los últimos años, se ha enfocado la atención en los anticuerpos anti-MOG, tras su descubrimiento en varios subgrupos de pacientes con EID, como trastornos del espectro neuromielitis óptica (del inglés NMOSD), encefalomielitis aguda diseminada (ADEM) y EM 9. Desde entonces se han desarrollado técnicas de detección con creciente sensibilidad y se han estudiado poblaciones más amplias, lo que ha permitido identificar fenotipos clínicos que hoy día son considerados típicos de MOGAD. En esta revisión se discutirán aspectos sobre físiopatología, epidemiología, manifestaciones clínicas y radiológicas, además del diagnóstico y el tratamiento.
Fisiopatología e inmunología
La MOG es un componente minoritario de la mielina que se expresa de forma selectiva en el SNC. Su función no se conoce por completo, pero parece actuar como una molécula de adhesión celular, regular la estabilidad de microtúbulos y modular interacciones inmunológicas de la mielina 10. Dado que esta glicoproteína se localiza en la superfície del oligodendrocito, es fácilmente accesible para anticuerpos IgG que han demostrado ser altamente inmunogénicos 10. A pesar de que la mayoría de los anticuerpos contra MOG humana no reconocen MOG de roedores, lo que dificulta su estudio 11, por medio de modelos animales de encefalomielitis autoinmune experimental se ha demostrado que los anticuerpos anti-MOG son directamente patogénicos, y ello induce la desmielinización 12) y conduce al depósito de complemento 13. Los estudios de patología han revelado la coexistencia de desmielinización perivenosa y cortical 14) y se describe una reacción inflamatoria mediada por linfocitos T CD4+ con inflamación granulocítica y depósito de complemento 14.
Epidemiología
La prevalencia de MOGAD es desconocida, pero algunos estudios la estiman entre 1,12 y 2,5 casos por cada 100.000 habitantes 15,16. La MOGAD puede aparecer desde la infancia hasta la vida adulta, con un pico entre los 30 y los 40 años. Existen diferencias en las manifestaciones clínicas según la edad de presentación, siendo más frecuente el debut antes de los 20 años en forma de ADEM, entre los 20 y los 30 años con neuritis óptica (NO), y con mielitis entre los 40 y los 50 años 17. No parece haber una predominancia por el sexo femenino tan marcada como en otras EID, con una relación hombre: mujer de 1:1,2 18.
Manifestaciones clínicas y fenotipos
Dada la similitud entre sus manifestaciones y las de NMOSD, anteriormente se incluía en esta última clasificación, pero en la actualidad se sabe que son entidades independientes 19. No obstante, debido a sus similitudes en relación con las topografías comprometidas en el SNC, es fundamental conocer las principales claves que diferencian estas entidades entre sí y de la EM, como se muestra en la tabla 1. MOGAD se presenta con diferentes fenotipos clínicos. El curso puede ser monofásico o recurrente y no se ha demostrado una progresión clínica entre recaídas 17,20. El pronóstico a largo plazo parece no diferir de forma significativa en comparación con el de NMOSD 21, aunque existe muy poca evidencia al respecto. A continuación se discuten las formas de presentación clínica más frecuentes.
Tabla 1 Características demográficas y clínicas de MOGAD, NMOSD y EM
Fuente: elaborac¡ón prop¡a.
Neuritis óptica
La NO es la presentación más frecuente de MOGAD en adultos. Con una frecuencia de entre 54 y 61 % 22, la afección visual suele ser grave al momento de alcanzar el nadir, está comúnmente asociada a papilitis, y tiende a comprometer el nervio óptico de manera bilateral 23. El riesgo de recurrencia es alto tras el primer episodio, con una mediana de 44 meses hasta la primera recurrencia 17. Una diferencia clave con la NO en NMOSD es el pronóstico visual, que es generalmente peor en ésta última 24. Sin embargo, se han descrito secuelas visuales en hasta el 80 % de los casos 17,25. La neuropatía óptica inflamatoria crónica recurrente (del inglés CRION) está asociada a la presencia de anticuerpos anti-MOG 26. Esta forma de presentación se caracteriza por episodios recurrentes de NO, buena respuesta al tratamiento con corticoides y dependencia de estos 27. Esto último está demostrado por la recurrencia de NO, que se presenta en promedio a los dos meses siguientes a la suspensión o el descenso de la dosis de corticoides 26.
Mielitis
Cerca de la mitad de los pacientes con MOGAD tendrán ataques de mielitis en algún momento del curso de la enfermedad 28. La afección de los esfínteres es de especial importancia, ya que existe una mayor tendencia a afectar los segmentos más bajos de la médula torácica y el cono medular 17. Si bien el compromiso residual de la función de los esfínteres es bastante frecuente, la minoría de los pacientes tendrá una discapacidad permanente relacionada con estos síntomas 17.
En el curso posterior a una mielitis longitudinalmente extensa en MOGAD, las recurrencias corresponden a una NO en un 70 % de los casos 17. Esto genera dificultades al momento del diagnóstico diferencial, por superposición con los criterios de NMOSD sero-negativa para anti-acuaporina 4 (AQP4) 29.
Encefalomielitis diseminada aguda
La ADEM es una EID que típicamente se presenta como un proceso monofásico asociado con síntomas de disfunción neurológica multifocal con encefalopatía, y que aparece más frecuentemente en edades pediátricas 30. Por mucho tiempo, la ADEM fue considerada una enfermedad idiopática, pero recientemente se ha encontrado que hay una alta seroprevalencia de anticuerpos anti-MOG, que oscila entre 19 % 31 y 58 % cuando se han utilizado técnicas de laboratorio más sensibles 32,33. En adultos, los casos de ADEM asociados a anticuerpos anti-MOG suelen cursar con crisis epilépticas 34. Los pacientes con anticuerpos anti-MOG tienen alto riesgo de un segundo episodio desmielinizante tras un ADEM, con tasas de recurrencia de hasta 88 % en pacientes persistentemente seropositivos 32,33,35.
Tallo cerebral
El compromiso del tallo cerebral está presente en alrededor del 30 % de los pacientes con anticuerpos anti-MOG que cursan con NO y/o mielitis 23. Sin embargo, también puede presentarse en forma de lesiones aisladas, con manifestaciones clínicas variadas que incluyen oftalmoplejía internuclear, disartria, compromiso respiratorio, disfagia, ataxia o un síndrome del área postrema. Estas son similares, pero ocurren con menor frecuencia que las descritas en NMOSD por anticuerpos anti-AQP4 17.
Encefalitis no-ADEM
MOGAD también se puede manifestar en forma de encefalitis que no satisface criterios de ADEM. Esto ocurre en el 20-30 % de los pacientes que se presentan con síndromes encefálicos 36,37, y suele manifestarse con crisis epilépticas, cambios comportamentales, trastornos del movimiento e incluso fiebre. Cerca de la mitad de estos casos puede tener compromiso cortical extenso y el 10 % puede tener un síndrome meníngeo 36. Se estima que el 10 % de los pacientes con clínica sugestiva de encefalitis autoinmune puede tener anticuerpos anti-MOG en suero como causantes o contribuyentes de los síntomas 38. El perfil clínico de estos pacientes puede ser de encefalitis cortical o límbica 38. El 80 % de los pacientes puede cursar con crisis epilépticas o fallas mnésicas, y la mitad puede presentarse con disminución del nivel de alertamiento 38. La cefalea y la fiebre también pueden ser síntomas muy frecuentes 37,39. Cerca del 10 % de los pacientes con MOGAD que se presentan con encefalitis no-ADEM puede tener una encefalitis autoinmune concomitante por anticuerpos contra el receptor NMDA (NMDAr), y puede presentarse con manifestaciones clínicas similares a las descritas anteriormente 40. Parece ser que los casos de encefalitis mediada por anticuerpos anti-MOG tienen mayor riesgo de recurrencias que aquellos sin estos anticuerpos, incluso si hay anticuerpos anti-NMDAr 38. Así mismo, el perfil clínico puede influir en el riesgo de recurrencias, siendo mayor para el fenotipo de encefalitis cortical en comparación con el de encefalitis límbica 38.
Diagnóstico
Diagnóstico serológico
Poco después de su descubrimiento, MOG fue relacionada con encefalitogénesis en la encefalomielitis alérgica experimental 41. Sin embargo, pronto se reconoció que los ensayos utilizados para detectar MOG por medio de ELISA generaban un gran número de falsos positivos 42. Más tarde se concluyó que estos resultados obedecían a la detección de epítopes de las porciones de MOG que no están expuestos al sistema inmune de forma natural 31.
El advenimiento de las técnicas de análisis basadas en células (del inglés CBA) ha permitido incrementar la sensibilidad y especificidad para detectar anticuerpos anti-MOG, lo que a su vez ha facilitado la configuración de los síndromes clínicos descritos anteriormente. Este tipo de pruebas utilizan células de líneas embrionarias de riñón u ovario de hámster chino (del inglés HEK y CHO, respectivamente) obtenidas por medio de cultivo, que son transfectadas con el gen de MOG, y la expresan en su configuración natural, lo que permite su detección 31. Estas células son expuestas al suero o líquido cefalorraquídeo del paciente sospechoso y posteriormente a anticuerpos secundarios dirigidos contra lgG1 12, que son detectados por medio de citometría de flujo o inmunofluorescencia indirecta. Existen varios CBA que han demostrado una alta tasa de acuerdo entre sí 43.
La persistencia de anticuerpos anti-MOG se ha asociado con una enfermedad de peor pronóstico, mientras que la presencia transitoria de anti-MOG se caracteriza por un curso monofásico. Esto sugiere que los anticuerpos anti-MOG pueden ser biomarcadores de respuesta al tratamiento, y por esta razón se recomienda hacer análisis serológico seriado después de 6 meses del inicio de los síntomas 12.
Diagnóstico por imágenes
En MOGAD se pueden identificar diferentes manifestaciones radiológicas en nervios ópticos, cordón espinal y parénquima cerebral, por lo que su caracterización imagenológica debe realizarse con resonancia magnética contrastada del neuroeje. Algunas de las manifestaciones radiológicas de MOGAD permiten diferenciarla de otras EID como EM y NMOSD.
Éstas cuales se presentan a continuación, de acuerdo con el segmento anatómico comprometido (figura 1).
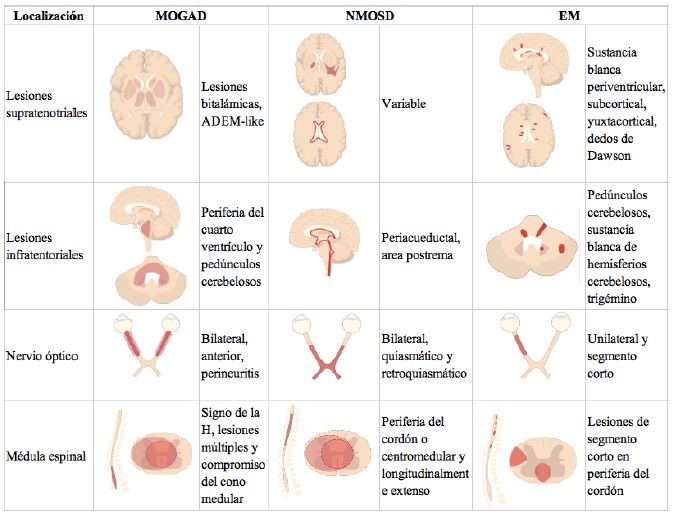
Fuente: adaptado de Dutra et al. 44, con autor¡zac¡ón expresa del t¡tular de derechos de autor, la Soc¡edad Rad¡ológ¡ca de Norte Amér¡ca (RSNA).
Figura 1 Representación esquemática de las características imagenológicas más comunes de las lesiones por enfermedad asociada a anti-MOG (columna izquierda), NMO (centro) y EM (derecha), en nervio óptico, médula espinal y encéfalo.
Nervios ópticos
La afección del nervio óptico en MOGAD tiene características radiológicas que pueden ser muy útiles para diferenciar la NO por esta entidad de otras causas. La afectación de los nervios ópticos por MOGAD ha sido típicamente descrita como bilateral y longitudinalmente extensa (> 20 mm) y de predominio prequiasmático. Adicionalmente, es frecuente identificar perineuritis y realce perineural postcontraste 45,46. Como diagnóstico diferencial, en NMOSD seropositiva predomina la afectación bilateral de la porción posterior de la vía óptica, incluido el quiasma y el tracto óptico. Las lesiones quiasmáticas son significativamente más frecuentes en NMOSD que en MOGAD 47. En estudios de seguimiento longitudinal se ha encontrado que casi la totalidad de los nervios afectados en MOGAD demuestran cambios involutivos 46.
Médula espinal
La hiperintensidad en T2/STIR de más de 2/3 de la circunferencia del cordón medular es conocida como patrón de mielitis transversa y constituye el principal hallazgo imagenológico en pacientes con lesiones del cordón espinal por MOGAD. En la fase aguda, la mayoría (70-80 %) de los pacientes se presenta con una lesión medular longitudinalmente extensa, cerca de 60 % puede tener dos lesiones o más, y lo más común es que se afecten simultáneamente tanto la médula cervical como la torácica 28,48. Aunque es usual la presencia de edema con aumento del volumen medular, no suelen encontrarse signos de necrosis 48. Si bien la localización de las lesiones y su topografía puede ser muy similar a las encontradas en los pacientes con NMOSD 49,50, hay rasgos distintivos de la mielitis por MOGAD, entre los que se resalta la afección del cono medular 49 y la presencia de hiperintensidad en el plano axial de la sustancia gris medular, lo que se conoce como "signo de la H" 28. En contraposición, en EM y NMOSD, el epicentro de la hiperintensidad suele encontrarse en sustancia blanca en la periferia del cordón -por fuera de la "H"- 28.
Adicionalmente, en MOGAD es usual encontrar lesiones en múltiples segmentos medulares, siendo infrecuente el realce postcontraste de estas 28,48. A diferencia de lo descrito, en EM predominan lesiones de segmento corto y en la periferia del cordón, con realce posterior a la administración de medio de contraste.
Cerebro
En alrededor del 10 % de los casos, las lesiones encefálicas en MOGAD pueden satisfacer criterios de diseminación en tiempo y espacio de EM 51. Los hallazgos cambian de acuerdo con la edad de presentación: en niños predominan las lesiones asimétricas hiperintensas en T2 de localización supra e in-fratentorial -consideradas típicas de ADEM- 52, así como lesiones bilaterales en los tálamos 53; en adultos suele identificarse una menor cantidad de lesiones cerebrales, que típicamente presentan una disposición con base cortical o localización in-fratentorial, en la periferia del cuarto ventrículo o en pedúnculos cerebelosos 20. Estas lesiones son característicamente mal definidas y de aspecto algodonoso 20. Las lesiones por MOGAD no suelen comprometer el área postrema, y por esto el identificar respeto de esta estructura, favorece el diagnóstico de MOGAD sobre el de NMOSD 54.
En el subgrupo de pacientes con encefalitis no-ADEM es posible identificar cambios imagenológicos propios de encefalitis autoinmune, caracterizados por aumento en el grosor cortical e hiperintensidad en T2/FLAIR de lóbulos temporales mesiales y sistema límbico, así como en corteza extralímbica de manera unilateral o bilateral asimétrica 37. En estos pacientes es infrecuente observar restricción a la difusión o hemorragia, y puede identificarse realce giriforme e incluso meníngeo adyacente 36,50. La presencia de anormalidades en la resonancia cerebral se ha asociado con una mayor discapacidad, y con la presencia de bandas oligoclonales y pleocitosis en el líquido cefalorraquídeo 20.
Criterios diagnósticos
Recientemente se han propuesto criterios diagnósticos de la MOGAD, por parte de un grupo internacional de consenso de expertos 55. Estos requieren la satisfacción de tres condiciones: A) la presencia de un síndrome desmielinizante típico de MOGAD (denominado como manifestación nuclear, como los que se describen arriba); B) la presencia de anticuerpos anti-MOG documentados a través de una prueba CBA; y C) la exclusión del diagnóstico de EM, que característicamente depende del juicio clínico experto 55. Estos pueden ser soportados por hallazgos característicos en las imágenes por resonancia, como se describieron anteriormente.
Tratamiento
Las recomendaciones terapéuticas para el manejo de la MOGAD se derivan en su mayoría de la experiencia adquirida en el manejo de la NMOSD seropositiva, y no se basan en evidencia de ensayos clínicos aleatorizados, que solo se han iniciado hasta el año 2022 con rozanolixizumab 56 y satralizumab 57. El tratamiento de MOGAD se basa en dos decisiones terapéuticas cardinales: el manejo de los ataques agudos y la decisión de establecer una terapia de mantenimiento que tiene como objetivo evitar las recurrencias y la acumulación de discapacidad a largo plazo 22.
Tratamiento de los ataques agudos
El tratamiento de primera línea en las recaídas es el uso de dosis altas de corticoides 58. En algunos casos, los corticoides no son efectivos, razón por la cual se recomienda se recomienda una segunda línea con plasmaféresis o inmunoglobulina humana intravenosa (IgIV) 48. Sin embargo, no existe evidencia sólida para la implementación de esta estrategia, razón por la cual la decisión debe basarse en la situación clínica del paciente y el criterio clínico.
Corticoides
El esquema terapéutico más común es el de la metilprednisolona endovenosa en dosis de 20 a 30 mg/ kg/día en niños y de 1 g cada día por 3 a 5 días para adultos 22. Aunque cerca de la mitad de los pacientes puede tener recuperación incompleta, la mejoría en respuesta a los corticoides suele ser rápida. Adicionalmente, existe una tendencia a recurrir tras la retirada de los corticoides 59.
El retiro progresivo de los corticoides tras un periodo de aproximadamente 3 meses después de la recaída puede ayudar a mitigar el riesgo de recurrencias, tanto en niños como en adultos. Se puede indicar con prednisolona oral, iniciando con 1 mg/kg/día (máximo 60 mg/día) y disminuyendo a 0,5 mg/kg/día durante hasta 4 semanas. Reducciones ulteriores deben hacerse más lentamente hasta completar al menos 3 meses postrecaída 58,60. Algunos estudios han mostrado un riesgo de recurrencias de hasta 60 % cuando se hace la disminución en menos de tres meses 25, lo que avala la indicación de la retirada lenta. Un subgrupo de pacientes puede mantenerse libre de recaídas sin requerir tratamiento inmunosupresor a largo plazo, razón por la cual los corticoides pueden ser la única terapia necesaria después del primer ataque 48.
Plasmaféresis
La plasmaféresis constituye la segunda línea de tratamiento en las recaídas de MOGAD y se indica en caso de resistencia a los corticoides o cuando exista una contraindicación para su uso. La dosis recomendada es de 3 a 5 recambios plasmáticos, que se hacen según la evolución clínica 22,48. La plasmaféresis ha sido estudiada como tratamiento de primera línea en MOGAD, y se ha encontrado que el 40 % de los pacientes mejoran por completo tras 5 recambios plasmáticos en promedio 48. Se desconoce aún si se puede considerar el uso simultáneo de corticoides y plasmaféresis como terapia de primera línea.
IgIV
Algunos estudios y reportes de casos soportan el uso de IgIV en recaídas graves de MOGAD o como alternativa en casos refractarios a los esteroides 20. Se propone también como una terapia modificadora de la enfermedad a largo plazo, sobre todo en niños. La dosis propuesta en recaídas es de 2 g/kg distribuidos en 5 días 61.
Terapia de mantenimiento
A la fecha no existen parámetros definidos que permitan predecir si los pacientes con MOGAD tendrán recaídas en el futuro, pero cada vez es más claro que quienes debutan en edad adulta tienen mayor probabilidad de discapacidad y recurrencias 17. Por tanto, es imprescindible establecer un tratamiento, teniendo en cuenta el balance individual de riesgo-beneficio, y determinar además factores como la gravedad y el grado de recuperación de las recaídas y otros que pueden predecir las recurrencias, como la seropositividad anti-MOG persistente 62. En la actualidad, el uso de las terapias de mantenimiento para evitar las recaídas del MOGAD se ha derivado de los regímenes terapéuticos empleados en NMOSD. La evidencia de la que disponemos viene principalmente de estudios observacionales y retrospectivos. Los medicamentos principalmente empleados en MOGAD son micofenolatomofetilo, azatioprina, IgIV y rituximab 25,48. Es importante tener presente que hasta el 75 % de los pacientes con MOGAD tratados con inmunosupresores continúan presentando recaídas a pesar de su uso 60.
En la tabla 2 se resumen los principales inmuno-supresores indicados para evitar recurrencias, de acuerdo con la evidencia disponible en la actualidad así como su modo de uso 25,48,63-66.
Tabla 2 Inmunosupresores de mantenimiento con evidencia de uso en MOGAD
| Inmunosupresor | Dosis, frecuencia, vía administración | Monitorización | Efectos adversos | Evidencia de uso y recomendaciones |
|---|---|---|---|---|
| Azatioprina | 2-3 mg/kg/día, oral | Hemograma, Transam¡nasas, TPMT* | Supres¡ón médula ósea, r¡esgo de ¡nfecc¡ones, hepatotox¡c¡dad, superv¡sar neoplas¡as (l¡nfoma y cáncer de p¡el) |
-ARR: 1,5 vs. 0,43, recaída 45 % 64 -ARR: NA, recaída 85 % (32 % < 3 m, 9 °/o vs. 3-vs. 6 m) después ¡n¡c¡o 63 -ARR: 1,4 vs. 0,43, recaídas: 64 % 48 - Se recom¡enda tomar en conjunto con cort¡co¡des orales |
| Micofenolato mofetilo | 600 a 1200 mg/m2/día (máx. 2 g/día) Oral | Hemograma, transam¡nasas |
Supres¡ón médula ósea, leucopen¡a, r¡esgo de ¡nfecc¡ones. Síntomas gastro¡ntest¡nales: náuseas y d¡arrea S¡ se usa a largo plazo, v¡g¡lar el r¡esgo de neoplas¡as. Alto r¡esgo de teratogen¡c¡dad |
-ARR: 1,20 vs. 0,23, recaída 27 % 64 -ARR: 0,9 vs. 0, recaída: 73 % 63 - Se recom¡enda tomar en conjunto con cort¡co¡des orales |
| Inmunoglobulina humana |
Intravenosa: Inducción: 1-2 g/kg x 5 días (máx. 1g/kg/día) Mantenimiento: 1 g/kg en 3 días Mensual. Subcutánea: 0,5-0,8 g/kg mensual |
Hemograma, BUN, creat¡n¡na | Cefalea, náuseas, artralg¡as y/o m¡alg¡as, reacc¡ón alérg¡ca a la ¡nfus¡ón. |
En adultos: -ARR: 1,0 vs. 0,1, recaída: 20 % 63 En niños: -ARR: 2,16 vs. 0,51, recaída 33 % 65 -ARR: 2 vs. 0 recaída: 71% 25 |
| Rituximab |
Inducc¡ón: 375-500 mg/m2 con ¡ntervalo de 2 semanas (máx. 1 g/¡nfus¡ón ¡ntravenosa) Manten¡m¡ento: 375500 mg/m2 (máx. 1 g/ ¡nfus¡ón) Cada 6 meses, ¡ntra-venosa o s¡ L¡nfos B CD19+ > 10x106 células/L < 6 meses |
Hemograma, transam¡-nasas, L¡nfos B CD19+, ¡nmunoglobul¡nas IgG, IgM, IgA | Reacc¡ón alérg¡ca a la ¡nfus¡ón: prur¡to, cefalea, rash o fiebre. Infecc¡ones severas, leucopen¡a pers¡stente o h¡pogammaglobul¡-nem¡a. |
-ARR: 1,08 vs. 0,43, recaída 27 % 64 -ARR: 2,4 vs. 0,59 recaída 57 % 63 -ARR: NA, recaídas: 78 % 48 -ARR: NA, recaídas: 38 % 66 |
Nota: *Genot¡p¡ficac¡ón TPMT (t¡opur¡na met¡ltransferasa). ARR: tasa anual¡zada de recaídas; se muestra la ARR pretratam¡ento (# recaídas por año antes del tratam¡ento, excluyendo el evento índ¡ce) vs. promed¡o ARR en tratam¡ento (# de recaídas por año en un mín¡mo de 6 meses de tratam¡ento). NA: no apl¡ca.
Conclusiones
MOGAD es una entidad independiente que hace parte de las EID del SNC. Afecta tanto a niños como a adultos, con características clínicas y radiológicas diferenciales. Su diagnóstico se basa en la sospecha clínica, la búsqueda de hallazgos radiológicos en resonancia magnética del neuroeje y la realización de la prueba serológica con técnica basada en células para la determinación de anticuerpos IgG anti-MOG. No existe evidencia sólida para emitir recomendaciones terapéuticas, pero MOGAD parece responder a la inmunoterapia convencional.

