











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
Permalink
Introducción
El síndrome de Leigh (OMIM#256000, SL) es una rara enfermedad neurodegenerativa, potencialmente mortal, causada por mutaciones de genes en las proteínas codificadas por el genoma nuclear o mitocondrial en la vía de la fosforilación oxidativa (OXPHOS), las cuales ocasionan una disfunción del metabolismo energético mitocondrial 1,2. Afecta a alrededor de 1 de cada 30.000 a 40.000 recién nacidos 2,3.
Su primera descripción data de 1951 por el neuropatólogo Denis Archibald Leigh, quien hizo una correlación anatomopatológica y un análisis post mortem en pacientes con encefalopatía subaguda de curso severo, progresivo y letal; en 1977 se hizo su primera asociación con disfunción de la cadena respiratoria, y posteriormente se publicaron artículos que completaron la descripción clínica de un síndrome neurodegenerativo. La tomografía computarizada (TC) y la resonancia magnética (RM) cerebral muestran lesiones núcleo basales características y el tallo cerebral, y los estudios genéticos permiten identificar las variantes patogénicas en el ADN mitocondrial y nuclear 1.
El diagnóstico del SL se fundamenta en la identificación de tres características distintivas: la presencia de una enfermedad neurodegenerativa, la disfunción mitocondrial por un defecto genético y la presencia de lesiones bilaterales simétricas en el sistema nervioso central mediante estudios de neuroimagen 3.
Caso clínico
Lactante de sexo femenino que ingresa a los 5 meses, nacida de padres no consanguíneos, producto de la primera gestación controlada, tamizaje infeccioso negativo, nacimiento a término, con adaptación neonatal espontánea y antropometría normal. No tenía antecedentes familiares de enfermedades neurodegenerativas. El neurodesarrollo fue normal hasta los tres y medio meses de vida, luego tuvo regresión de hitos del desarrollo, con pérdida del sostén cefálico y la sonrisa social, disminución de los movimientos espontáneos e hipoalimentación. Al ser valorada en el servicio de urgencias se evidenció microcefalia, somnolencia, llanto débil, hipotonía, sin seguimiento visual de objetos, reflejos musculotendinosos normales, sin rasgos dismórfícos.
Los paraclínicos del ingreso mostraron acidemia metabólica con aumento del anion GAP, hiperamonemia leve (en 90 umol/L, normal inferior a 75 umol/L) y lactato elevado (5,8 mmol/L, normal inferior a 2 mmol/L). Se sospecha una enfermedad neurometabólica de tipo aciduria orgánica primaria debido a la presencia de acidemia metabólica hiperlactatémica e hiperamonemia leve. Además, en la TC simple de cráneo se observó hipodensidad bilateral en el cuerpo estriado y en la parte anterior del cíngulo derecho, sugestivo de encefalopatía necrotizante subaguda y SL. Se inicia administración de cofactores: riboflavina, tiamina, L carnitina, coenzima Q10, ácido α-lipoico y biotina. Las imágenes de RM cerebral simple con espectroscopia mostraron lesiones de aspecto necrotizante en núcleos basales con compromiso de núcleos caudados, putámenes y tálamos, edema citotóxico cortical, espectroscopía con aumento de la colina (Cho), disminución del N acetil aspartato (NAA) y pico de lactato, hallazgos característicos de la espectroscopia por RM en SL (figuras 1 y 2).
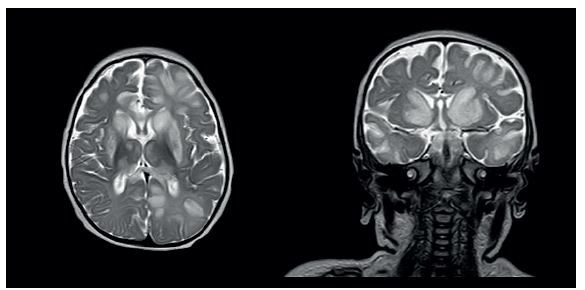
Fuente: elaboración propia.
Figura 1 Resonancia magnética secuencia T2: A y B en corte coronal y axial a la altura de los núcleos de la base, donde se evidencian hiperintensidades que comprometen la cabeza de los núcleos caudados, putámenes y talamos. Adicionalmente, se presentan lesiones hiperintensas en la sustancia blanca subcortical de los lóbulos frontales de predominio izquierdo, así como en el lóbulo occipital y temporal izquierdo.
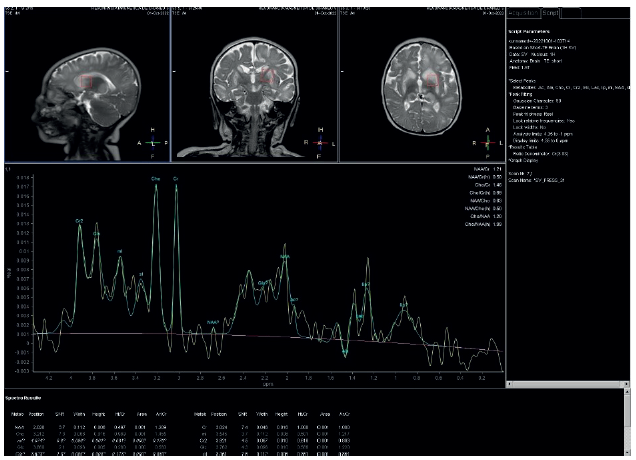
Fuente: elaboración propia.
Figura 2 Espectroscopia univoxel tiempo de eco corto localizada entre los núcleos caudado y putamen izquierdos que muestra elevación de la colina, disminución de NAA y pico de lactato, hallazgos secundarios a necrosis.
En valoración por oftalmología se encontró atrofia óptica bilateral, con atrofia vascular y de la capa de fibras nerviosas; los aminoácidos séricos tenían elevación de alanina, los ácidos orgánicos en orina lactaturia y la secuenciación genómica completa reportó una mutación homocigota con cambio de sentido p.ser270Gly (c.808A>G) en el gen HIBCH (OMIM®: 250620), sugestiva de deficiencia de 3-hidroxiiso-butril-CoA hidrolasa HIBCH. Se hizo asesoramiento genético y consejería reproductiva porque el modo de herencia de la enfermedad es autosómica recesiva, pero los padres de la paciente no desean el estudio genético para identificar el estado de portador por paternidad satisfecha.
Posteriormente, tuvo varios ingresos a hospitalización por deterioro clínico debido a infecciones febriles agudas de etiología viral. A los 7 meses de vida inició con crisis epilépticas motoras bilaterales no provocadas, por lo que se inició levetiracetam.
En staff de neurología se discutió el caso de la paciente y se sugirió la administración de formula exenta de valina, pero no fue posible la administración porque en Colombia las fórmulas nutricionales exentas de este aminoácido están autorizadas exclusivamente para pacientes afectados por enfermedad de la orina con olor a jarabe de arce, acidemia metilmalónica o acidemia propiónica.
La paciente tuvo recurrencia de las crisis epilépticas a los 10 meses, que fueron presenciadas por neurología y clasificadas como espasmos epilépticos flexores. El electroencefalograma (EEG) en vigilia mostró patrón de hipsarritmia, sin foco dominante. Se inició tratamiento con vigabatrina a 200 mg/kg/día, pero a las 24 horas inició con apneas de 6 segundos de duración y persistieron los espasmos epilépticos, por lo cual se suspendió este anticonvulsivo. A las 96 horas la paciente continuó con empeoramiento respiratorio y clínico, tiempo en que se esperaba que los niveles de vigabatrina fueran insignificantes, con base en su farmacocinética. Los reactantes de fase aguda y los rayos X de tórax anteroposterior y lateral fueron normales. Se acompañó a la familia y a la paciente, en conjunto con cuidado paliativo y psicología. Luego, la paciente falleció por insuficiencia respiratoria aguda, sin signos de sufrimiento clínico.
Discusión
El SL es una encefalopatía mitocondrial que ocurre principalmente en lactantes y niños menores antes de los 2 años de vida. Es reconocida como la presentación clínica pediátrica más común de las enfermedades mitocondriales. Comprende diferentes tipos de transmisión, ligada al cromosoma X, autosómica recesiva y mitocondrial/materna, de acuerdo con la ubicación del genoma nuclear o mitocondrial alterado o en la interacción entre los dos genomas. Las alteraciones genéticas explican parcialmente la gran heterogeneidad y el amplio rango de presentación clínica 2,4,5.
El inicio de las manifestaciones clínicas se da después de un período inicial de desarrollo normal; también se ha reportado inicio congénito hasta en el 22 % de los casos 6. Las manifestaciones neurológicas incluyen rápida disminución de las funciones cognitivas y motoras, regresión del desarrollo, hipotonía muscular, ataxia, distonía, convulsiones, anomalías oftalmológicas como nistagmo, atrofia óptica, ceguera y apnea. Las manifestaciones no neurológicas incluyen falla de medro, microcefalia, anomalías cardíacas, renales, hepatológicas y hematológicas 3.
El SL tiene habitualmente un curso rápidamente progresivo y la mayoría de los niños mueren antes de cumplir los 3 años 2,4,7. Aunque también se ha reportado SL de inicio en adultos como una presentación muy rara, observada en las variantes patogénicas en el gen MTHFR-metilentetrahidrofolato re-ductasa, uno de los genes nucleares más frecuentes como causa de SL que puede tener un inicio tardío, hasta los 17 años, curso más leve y con supervivencias en edades adultas 8.
Entre los principales diagnósticos diferenciales de SL tenemos la encefalomiopatía mitocondrial, con acidosis láctica y episodios parecidos a un accidente cerebrovascular, síndrome Alpers-Huttenlocher, la ataxia sensorial con miopatía epiléptica mioclónica, el espectro de neuropatía atáxica, síndrome de ME-GDEL, acidosis láctica congénita, síndrome de Pearson y síndrome de Sengers 9.
La primera mutación génica involucrada en SL se identificó en 1991. Desde ahí se han reconocido más de 75 genes mutados, con diferentes modos de herencia, de los cuales el 70 % al 90 % pertenece a genes ubicados en el núcleo celular y el restante 10 % al 30 % tiene que ver mutaciones en el ADN mitocondrial 3,10. Estos en su mayoría codifican proteínas de la vía OXPHOS, genes involucrados en la replicación, la transcripción y la traducción del DNA mitocondrial, otras enzimas relevantes como piruvato deshidrogenasa y proteínas de la biosíntesis de la coenzima Q10, la oxidación de ácidos grasos y procesos no mitocondriales como el metabolismo de la tiamina con afectación indirecta de la función mitocondrial 2,7. Se desconoce la causa genética de al menos el 50 % de los casos de pacientes 6.
La deficiencia del complejo I, junto con la deficiencia del complejo IV, son las causas más comunes de trastornos de la cadena respiratoria mitocondrial, y entre el 35 % y el 50 % son diagnosticados con SL. Otra alteración poco común en SL es la alteración de la vía de degradación de la valina, en la cual la L-valina se convierte en succinil-CoA, y aumenta secundariamente la concentración de valina y su toxicidad. Se han identificado mutaciones en dos genes: HIBCH y ECHS1, las cuales ocasionan múltiples deficiencias del complejo de la cadena respiratoria mitocondrial y deficiencia del complejo piruvato deshidrogenasa 6,10.
En el caso presentado identificamos la mutación del gen nuclear mitocondrial HIBCH que codifica la proteína 3-hidroxiisobutiril-CoA hidrolasa, ubicado en el cromosoma 2q32. Esta enzima cataliza el quinto paso del metabolismo de la valina, responsable de convertir 3-hidroxiisobutril-CoA en ácido 3-hidro-xiisobutírico por el catabolismo de la valina, así como 3-hidroxipropanoil-CoA en ácido 3-hidroxipropió-nico, una ruta secundaria hipotética para el catabolismo del ácido propiónico, que alimenta la ruta catabólica de la valina 11,12.
La deficiencia de HIBCH tiene una incidencia estimada en la población general entre 1 en 127.939 en asiáticos orientales y 1 en 551.545 en europeos. No hay datos sobre la incidencia de esta deficiencia en Latinoamérica 11,13. Sin embargo, en el 2019 se publicaron los casos de dos pacientes colombianos afectados con esta mutación de la HIBCH, y se identificó la misma variante que tenía nuestra paciente, p.ser270Gly (c.808A>G) 13.
Esta mutación en HIBCH ocasiona episodios de cetoacidosis, niveles elevados de piruvato y lactato en el líquido cefalorraquídeo y regresión de los hitos del desarrollo en el transcurso del primer año de vida. En las imágenes por RM cerebral es típica la alteración de señal en la secuencia T2 de los ganglios basales en forma bilateral y tronco encefálico, diferentes grados de atrofia de la sustancia blanca, y pico de lactato determinado mediante espectroscopia 13. Este hallazgo neurorradiológico ha sido común en los tres casos colombianos reportados a la fecha. Más infrecuentemente, se ha descrito la afectación de la sustancia blanca cerebral, de la sustancia negra, del núcleo rojo, de la médula espinal y del cerebelo 5,7. Nuestra paciente falleció por causa directa de una insuficiencia respiratoria aguda y en la MRI cerebral se ha reportado afectación medular caudal de la médula espinal, hallazgo relacionado con disfunción del centro respiratorio 14.
Otros hallazgos de laboratorio para la deficiencia de HIBCH incluyen la elevación de 3-hidroxi-isobutiril-carnitina en el perfil de acilcarnitinas séricas, como consecuencia de la acumulación de 3-hidroxi-iso-butiril-CoA. Esta alteración se ha informado en casi todos los sujetos con deficiencia de HIBCH y en la actualidad se considera un marcador metabolómico de tipo diagnóstico 15.
Candelo et al. reportaron dos casos colombianos no relacionados de enfermedad neurodegenerativa sindromática por variantes nuevas en el gen HIBC 13. Los pacientes tenían hipotonía axial con hipertonía espástica de los miembros inferiores y uno de ellos presentaba epilepsia intratable. El análisis de acilcarnitinas plasmáticas fue normal, destacando el amplio espectro fenotípico de la enfermedad ocasionada por esta mutación.
La deficiencia de HIBC también se asocia con disquinesia paroxística, un trastorno de movimientos infrecuente con gran heterogeneidad fenotípica y genética. Robertie et al. describieron el caso de un paciente con hemidistonía paroxística derecha y otro con disquinesia paroxística y corea de ambas manos, inducida por el ejercicio, que tenían alteración de la señal palidal en la RM cerebral y compartían la mutación homocigota c.913A > G, p.Thr305Ala en el gen HIBC 16.
En la actualidad, el tratamiento es principalmente sintomático e implica ajustes nutricionales. Como producto de ensayos clínicos e informes de casos se recomiendan algunos tratamientos paliativos para el control de los síntomas y con la intención de restablecer las vías metabólicas relacionadas con la depuración de los metabolitos neurotóxicos 9. Se han propuesto diversas alternativas de tratamiento para pacientes con deficiencia de HIBCH, como basar la fuente principal de energía en los pacientes con carbohidratos y complementar su dieta con bajos niveles de valina y suplementos de carnitina, lo que podría reducir la producción de metacrilil-CoA en las células neuronales. También se ha empleado dieta cetogénica, terapia con reacción anaplerótica y que puede mejorar la función mitocondrial. No obstante, ninguna de estas intervenciones ha logrado prevenir la progresión de la enfermedad 5,13.
Conclusiones
El SL por mutación en el gen HIBCH es una enfermedad neurodegenerativa con presentación fenotípica y genotípica variable, caracterizado principalmente por pérdida de los hitos de desarrollo motor e intelectual, con patrón neurorradiológico y bioquímico característico. El diagnóstico de la enfermedad está basado en técnicas de biología molecular (secuenciación exómica o genómica). En este artículo mostramos que la enfermedad por mutación del HIBCH debe incluirse en el repertorio de los errores innatos del metabolismo con presentación severa, progresiva y con respuesta parcial a las opciones de tratamiento. El tratamiento actual contempla el alivio de los síntomas y modificación nutricional, con base en los trastornos metabolómicos ocasionados por la enfermedad, lo cual destaca el desafío y las limitaciones que implican los errores congénitos del metabolismo como posibles causas del SL.

