










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Odontología
Print version ISSN 0120-971X
CES odontol. vol.26 no.1 Medellìn Jan./June 2013
Bases genéticas de la formación de fisuras labiales y/o palatinas en humanos
Genetic basis of orofacial cleft formation in humans
Lina María Escobar,1 Jeanette Prada-Arismendy,2 Carolina Téllez,3 Jaime Castellanos4
1MSc. Facultad de Odontología. Universidad El Bosque. E-mail: linaescobar10@gmail.com.
2MD. MSc. Universidad El Bosque. E-mail: jpradaarismendy@yahoo.es.
3OD, Facultad de Odontología. Universidad El Bosque. E-mail: ctconti@gmail.com.
4OD, MSc, PhD. Facultad de Odontología. Universidad Nacional de Colombia. E-mail: jecastellanosp@unal.edu.co
Recibido: abril de 2013. Aprobado: junio de 2013
Resumen
La hendidura labio palatina (cleft lip/palate, CL/P) es una alteración craneofacial de alta frecuencia en la población mundial, cuya etiología es multifactorial, y que involucra factores tanto genéticos como ambientales. La mayoría de este tipo de patologías se presenta como un defecto no sindrómico (70%), y el resto de hendiduras se asocian con alteraciones adicionales en los defectos de tipo sindrómico. Gracias al desarrollo de herramientas moleculares se han identificado varios genes que están involucrados en CL/P de tipo no sindrómico, como son el proto-oncogen Bcl3, el gen Tgfb, el gen homeótico Msx1 y el gen Bmp entre otros, los cuales se han podido identificar mediante estudios de ligamiento y el uso de ratones deficientes en estos genes (knock-out). Por otra parte, se han identificado más de 300 síndromes diferentes asociados con CL/P, entre ellos uno de los más frecuentes el síndrome de van der Woude donde también se han identificado mutaciones en genes como el IRF6. Debido a que CL/P es una patología de alta complejidad etiológica donde las alteraciones genéticas juegan un importante papel y donde cada día se amplía el conocimiento sobre alteraciones en diversos genes que contribuyen a la formación de la alteración, el objetivo de este artículo, es revisar la información actualizada sobre la genética del CL/P y los genes reportados que pueden contribuir al desarrollo de esta compleja patología.
Palabras clave: Preservación del reborde alveolar, Reabsorción ósea, Extracción dental, Injerto óseo, Implantes dentales.
Abstract
Clefts of the lip and palate (CL/P) are a craneofacial alteration of high frequency in the world-wide population, whose etiology is multifactorial, involving genetic factors as much as environmental ones. Most of this type of pathologies (70%) appear as non-syndromic form, although the presence of additional facial alterations is associated with clefts of syndromic type. The development of molecular tools has permitted to identify some of the genes that are involved in non-syndromic CL/P, such as proto-oncogen Bcl3, gene Tgfb, homeotic gene Msx1 and Bmp among others, which have demonstrated a relationships between them using both linkage analysis and knock-out mice. In the other hand, they have been identified more of 300 different syndromes associated with CL/P, being one from most frequent the van der Woude syndrome which also have identified mutations in genes such as IRF6. Because CL / P is a highly complex etiology pathology where genetic alterations play an important role and where every day is more knowledge on alterations in several genes that contribute to the formation of the alteration, the aim of this article is review the current knowledge on the reported genetics of the CL/P and genes that would contribute to the development of this complex pathology.
Key words: Left lip/palate, Bone Morphogenetic Protein 2, Maxillofacial Development.
Introducción
La hendidura labiopalatina (CL/P, por su nombre en inglés cleft lip and palate) es el defecto congénito mas frecuente que afecta a las estructuras maxilofaciales del hombre, cuya etiología es bastante compleja, involucrando tanto factores genéticos como ambientales.1 Este defecto es frecuentemente encontrado en recién nacidos y por tanto, ha sido objeto de innumerables estudios en todo el mundo. Se ha informado que la prevalencia es 1:500 nacidos vivos en Europa y de 1:1000 en Estados Unidos2 y Colombia.3
Las hendiduras orofaciales se han clasificado en formas sindrómicas y no sindrómicas. Aproximadamente el 70% de las fisuras labiopalatinas son no sindrómicas con una contribución genética de 20 a 30%, por lo cual el estudio de su etiología y patogénesis son complejas y pobremente entendidas. Por otro lado, se han reportado más de 300 síndromes genéticos que son acompañados de CL/P, mientras que otros son debidos, ya sea a rearreglos cromosómicos o a la agresión con teratógenos.4
A pesar de la existencia de una gran cantidad de estudios e investigaciones sobre los eventos biológicos que regulan el desarrollo del labio y el paladar, siguen siendo poco conocidas las variables que desencadenan la formación de CL/P. En los últimos años y gracias al avance de las herramientas moleculares y de un mayor conocimiento en la genética poblacional, se han podido identificar varios genes que están involucrados en la formación y desarrollo orofacial, evaluando a su vez el papel que podrían tener en la formación del CL/P sindrómico y no sindrómico. Dado lo anterior, se hizo una revisión de la información actualizada sobre la evidencia existente de la participación de la genética y los genes reportados en la aparición de defectos y hendiduras labiopalatinas.
Embriogénesis del labio y paladar
Para entender cómo las alteraciones genéticas conducen a la formación de CL/P es importante conocer como se lleva a cabo la formación del paladar y del labio superior en estado embrionario y en condiciones normales.
Durante las etapas iniciales de formación del embrión, este presenta una depresión ectodérmica denominada estomodeo que está constituida por la cavidad bucal y la nasal aun sin separación entre ellas. La palatogénesis permite que el paladar recién formado establezca una división entre estas dos cavidades dando origen a la boca y la nariz.5
En los humanos, el paladar se forma entre la semana 8 a 12 de vida intrauterina y está constituido por el paladar primario y el paladar secundario. El paladar primario es la parte más pequeña y anterior del paladar que comprende la premaxila, que debe fusionarse con la parte posterior o paladar secundario, la cual se forma por fusión de los dos procesos palatinos.5 En la Figura 1, se muestra esquemáticamente la secuencia de eventos que ocurre durante la palatogénesis.
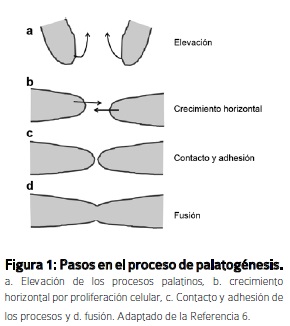
En el inicio de la palatogénesis los procesos palatinos aparecen como protrusiones en las paredes laterales de la cavidad oronasal y crecen verticalmente alrededor de la lengua. Posteriormente la elevación y el crecimiento de los procesos es producido por cambios y proliferación de las células mesenquimales derivadas de la cresta neural (Figuras 1a y b). Los procesos palatinos crecen hasta que el epitelio del borde medial de cada uno de ellos hace contacto con el proceso contralateral (Figura 1c). Una vez se establece el contacto entre los procesos se induce la eliminación por apoptosis del tejido epitelial y el crecimiento confluente de las células mesenquimales subyacentes7 (Figura 1d).
Por otra parte, las fisuras labiales se forman como espacios uni- o bilaterales entre el filtrum y la pared lateral del labio superior, a veces extendiéndose hasta el paladar secundario. El labio se forma a partir de la fusión de los procesos maxilar, medial nasal y lateral nasal. La fusión inicial se da entre los procesos nasales medial y lateral y posteriormente es seguido por la fusión de los procesos nasales medial y maxilar. Durante esta fusión los procesos se contactan, se adhieren y se produce una lamina epitelial media que desaparece rápidamente mediante apoptosis permitiendo la confluencia de las células mesenquimales y la finalización de la fusión.8
Cualquier falla en los pasos anteriormente enumerados conducirá a la formación de fisuras palatinas, labiales o la combinación de ambas.
Hendiduras labiopalatinas no sindrómicas
La mayoría de las CL/P (70%) son del tipo no sindrómico, es decir que el defecto se presenta sin otras anormalidades. Por análisis de ligamiento en familias con más de un individuo afectado, se han podido relacionar algunos loci con mayor susceptibilidad a la aparición de CL/P. Se ha reportado asociación entre CL/P y alteraciones en el gen de endotelina-1 (Et1) localizado en el cromosoma 6, el cual codifica para un péptido vasoactivo expresado en las células endoteliales vasculares. Estudios realizados en ratones knockout para dicho gen muestran hipertensión y defectos craneofaciales como reducción marcada del tamaño de la lengua, micrognatia y fisura palatina.9,10 Se ha establecido que el receptor para endotelina (Eta), es expresado en células ectomesenquimales derivadas de la cresta neural en los arcos braquiales y que alteraciones en su expresión y función produce defectos craneofaciales en ratones homocigotos deficientes Eta-/- 11.
Para otros genes, se han tenido resultados contradictorios en cuanto a su participación en la formación de CL/P no sindrómico. Por ejemplo mutaciones en el proto-oncogen Bcl3 localizado en el cromosoma 19, en un análisis de ligamiento en individuos con CL/P han mostrado desequilibrios de ligamiento, pero con un valor LOD (del inglés logarithm of odds) que no ha sido suficientemente alto para confirmar una relación entre el alelo y la patología. Por otra parte, no se ha podido asegurar tampoco que el gen para el receptor del ácido retinoico RARA localizado en el cromosoma 17 esté participando en una forma directa en la inducción de CL/P, sino que al parecer podría estar modificando la severidad de la patología.12,13
Estudios genéticos familiares en humanos de casos de CL/P no sindrómicos han mostrado que no hay un solo locus implicado en este tipo de alteraciones, lo que ha favorecido el desarrollo de un modelo multifactorial que se propone explicar el componente genético de este desorden.14 Sin embargo, estudios realizados en ratones han mostrado que la función anormal de un gen particular puede tener un papel importante en la etiología de las fisuras orales. La herramienta molecular de generación de ratones knock-out ha permitido definir algunos genes que pudieran estar asociados con la formación de fisuras labiopalatinas no sindrómicas. Algunos de estos genes se enuncian en la Tabla 1.

Dentro de los genes mencionados anteriormente se encuentra el gen Tgfb que pertenece a un grupo de moléculas de señalización intracelular importantes durante el proceso de desarrollo. La proteína TGFB3 se localiza de forma abundante en el epitelio de los procesos palatinos antes de que se lleve a cabo la fusión de los mismos y se ha visto que su unión y actividad a través de su receptor ALK-5 es indispensable para que ocurra la fusión de los procesos palatinos de forma adecuada.15
Se ha demostrado que los ratones knock-out para Tgfb3 presentan un fenotipo de fisura palatina y que la proteína TGFB3 controla el proceso de diferenciación de las células epiteliales a células mesenquimales en la parte medial de los dos procesos palatinos en ratones.12 Adicionalmente, se encontró que el tratamiento con proteína TGFB exógena es capaz de corregir los defectos en la fusión palatina en embriones de ratón TEta -/- 11 in vitro.16
Los miembros de la familia de Tgfb han sido involucrados en muchos procesos biológicos durante el proceso de palatogénesis como son la migración celular, la síntesis y depósito de matriz extracelular, la degradación de membrana basal, la proliferación de células mesenquimales y la apoptosis de células epiteliales del borde medial de las prolongaciones palatinas, para permitir la fusión. Debido a la importancia de TGFB para el desarrollo adecuado de la unión palatina embrionaria, las vías de señalización que involucran a TGFB han recibido gran atención en los años recientes.17
Para el caso del gen homeótico Msx1, el cual se expresa en los vertebrados en varios órganos durante el desarrollo, se ha encontrado evidencia que lo implica en la aparición de hendiduras. El gen Msx1 se expresa en el primordio facial, principalmente durante la organogénesis, en el sitio donde ocurren las interacciones entre células epiteliales y mesenquimales. Los ratones deficientes en el gen Msx1 mueren recién nacidos y presentan anormalidades craneofaciales severas incluyendo hendidura palatina, ausencia de procesos alveolares y detención del desarrollo dental.18
En humanos, se ha determinado la asociación entre mutaciones en el gen Msx1 con hendiduras palatinas no sindrómicas y agenesia dental, similar al fenotipo observado en los ratones Msx1 mutantes. En los ratones deficientes de Msx1 las prolongaciones palatinas bilaterales se forman y se elevan de forma normal pero falla en el establecimiento de su contacto y de la posterior fusión, dando formación a una hendidura.19
Adicionalmente, estudios tanto in vivo como in vitro han mostrado evidencias de que Msx1 controla vías de señalización que involucran a las proteínas morfogenéticas óseas (BMP) y Sonic Hedgegoh (Shh), que regulan el crecimiento del paladar durante la palatogénesis en mamíferos.20 Además de relacionarse con fisuras labiopalatinas no sindrómicas, Msx1 también se ha visto asociado con una serie de defectos craneofaciales incluyendo hendidura del paladar secundario, detención del desarrollo dental y anormalidades de varios huesos faciales.18
La asociación entre los genes (Msx1 y Tgfb) se ha determinado en niños suramericanos, pues se evidenció una asociación entre la variación genética en estos dos loci y la aparición de fisura labial con o sin paladar hendido y paladar hendido aislado en esta población, lo cual sugiere una posible interacción entre estos dos genes en el desarrollo de hendiduras orales.21 Por otro lado, un estudio realizado en 94 pacientes del proyecto Operación Sonrisa en Colombia cuyo objetivo era evaluar la asociación entre el desarrollo de hendidura labial no sindrómica con o sin formación de fisura palatina y 4 alelos polimórficos de repetición de dinucleótidos CA en Msx1, mostró una asociación estadísticamente significativa entre el alelo 3 del gen Msx1 y la presencia de hendiduras en una población colombiana.22
Teniendo en cuenta que las BMPs hacen parte de la superfamilia TGFB y que son ahora reconocidas como factores de crecimiento multifuncionales que participan en una gran variedad de funciones biológicas esenciales para la gastrulación, organogénesis, crecimiento embrionario y postnatal, se ha determinado que las vías de señalización mediadas por estas, están involucradas en el desarrollo y formación de una serie de elementos craneofaciales que incluyen la cresta neural craneana, el primordio facial, los dientes, los labios y el paladar.23
La señalización mediada por BMPs es un proceso importante para que se lleve a cabo la palatogénesis. Se observa un aumento del mRNA de BMP2, BMP3, BMP4 y BMP5 en el paladar de ratones, tanto en el epitelio como en el mesénquima, antes, durante y después de la fusión de las prolongaciones palatinas.24
La importancia de las BMPs en la palatogénesis se empezó a evidenciar mediante modelos de investigación con ratones a los que se les inducia hendidura palatina mediante aplicación de ácido retinoico y se asoció con expresión disminuida de BMP2, 4 y 5.24 Evidencia adicional se obtuvo mediante el estudio de ratones transgénicos con inactivación de Alk2 o Bmpr1a (receptores de BMPs), en los cuales se desarrollaron hendiduras orofaciales.23
Estudios en ratones knock-out para el receptor Bmpr1a, de mostraron que la formación del paladar secundario se asoció con una disminución en la proliferación celular y aumento en la apoptosis. Sin embargo, cuando las prolongaciones palatinas de los mutantes son colocadas juntas en cultivo, estas se fusionan, lo que quiere decir que la hendidura observada en ratones deficientes de Bmpr1a es debida a la falta de proliferación de células mesenquimales y no a una falla en la fusión del epitelio.23
Por otro lado, se encontró una elevada expresión de proteína BMP2 en el mesénquima adyacente a la línea media durante la fusión palatina, mientras que la función de BMP4 y 7 no fue esencial, como se demostró mediante ratones knock-out.25 Por lo tanto, estos estudios permiten concluir que la proliferación mesenquimal mediada por BMP2 es un evento clave en el proceso de palatogénesis.
Se ha propuesto que las BMPs se regulan durante los eventos de señalización inducidos por Msx1 durante la palatogénesis. La deleción de Msx1 conduce a hendiduras palatinas en ratones, y a su vez, la expresión transgénica de Bmp dirigida por el promotor de Msx1 conduce al rescate de la hendidura, lo cual sugiere que las BMPs están actuando corriente abajo de Msx1 en el proceso de la palatogénesis.20
Estos datos indican diferentes requerimientos de señalización de BMPs en la formación tanto del paladar como del labio. El desarrollo y la fusión del paladar son eventos relativamente tardíos en la embriogénesis y están influenciados de una forma crítica por el desarrollo de estructuras relacionadas. Todo esto hace de la palatogénesis un proceso bastante delicado que puede ser fácilmente afectado por factores genéticos y ambientales.
Hendiduras labiopalatinas sindrómicas
Se conoce que más de 300 síndromes se asocian con presencia de CL/P. Los casos sindrómicos de fisuras palatinas o labiopalatinas pueden ser subdivididas en aquellas que ocurren como parte de un desorden Mendeliano, aquellos que se originan a partir de anormalidades estructurales de los cromosomas, síndromes asociados a teratógenos conocidos o aquellos cuyas causa aun no han sido identificadas por lo cual no han sido caracterizados.12
Uno de los más comunes desórdenes autosómicos dominantes humanos asociado con CL/P es el síndrome de van der Woude (VWS, #119300) que corresponde al 1% de las casos de fisura labiopalatina sindrómica.26 Esta patología se ha asociado con la formación de hendiduras redondeadas o "pits" en la mucosa del labio inferior y con formación de CL/P.27 El locus para este síndrome ha sido previamente identificado en la región 1q32-q41 del cromosoma 1.28
Estudios genéticos han permitido la detección de 70 mutaciones sin sentido y cambios en el marco de lectura del gen del factor regulador del interferón IRF6, involucrado en la etiología del síndrome de van der Woude. Este gen codifica para un factor de transcripción que hace parte de una familia de 9 miembros involucrados en la regulación de la expresión del Interferón alfa y beta que ocurre después de la infección viral.29
El papel exacto de IRF6 durante el desarrollo embrionario orofacial no se conoce exactamente, sin embargo se ha detectado una alta expresión en diversas estructuras craneofaciales, incluyendo los bordes mediales de los procesos palatinos durante la fusión, los gérmenes dentales, los folículos pilosos y la piel. Se ha visto también que la haploinsuficiencia de IRF6 causa disrupción del desarrollo orofacial.29 Otros estudios han identificado mutaciones sin sentido en IRF6 asociados a CL/P aisladas sin ningún otro cambio fenotípico aparente.30
Por otro lado, el gen relacionado con el receptor del poliovirus PVRL-1 se ha establecido como responsable de la inducción del síndrome de displasia ectodérmica y CL/P (CLPED-1 #225060). Los análisis genéticos han mostrado mutaciones en la molécula de adhesión celular PVRL-1 que es altamente expresada en el desarrollo de la cara y del paladar. La mutación puntual W185X, genera riesgo moderado para CL/P dado que algunos de los individuos afectados en los estudios poblacionales, no portan dicha mutación. PVRL1 codifica para la molécula nectina-1, que permite la adhesión célula a célula y es expresada de forma significativa en la cara y el paladar en desarrollo.31
El síndrome autosómico dominante ectrodactilia, displasia ectodermal y hendidura orofacial (EEC, #129900) se caracteriza por defectos en manos y pies, anomalías en el cabello, dientes, glándulas sudoríparas, uñas y del conducto nasolacrimal y fisura labial con o sin fisura palatina. Se ha asociado con mutaciones heterocigotas en el gen p63 localizado en el cromosoma 3q27. Este gen es altamente expresado en el tejido ectodérmico y en los procesos maxilar y mandibular en el primer arco branquial.32 La mutación de este gen influencia la formación de hendiduras y las mutaciones en la región de unión al DNA produce ectrodactilia, displasia ectodermal y CL/P, mientras que la mutación en la región carboxilo terminal de la proteína producirá fisura labial o fisura palatina. Por su parte, las mutaciones en la región amino terminal aumenta el riesgo de presentar fisura palatina aislada.33 Actualmente se vienen estudiando moléculas de señalización corriente abajo de p63 como Jagged2 cuya expresión alterada induce en ratones la formación de fisuras palatinas.34
Otro de los defectos genéticos que se caracteriza por presentar CL/P pero además inducir alteraciones en las estructuras mediofaciales corresponde al síndrome de Opitz (GBBB #300000) que se asocia con mutaciones en el gen MID1 localizado en el cromosoma Xp22.35
El desorden de fisura palatina ligada al cromosoma X, (CPX #303400), es un raro desorden caracterizado por presentar fisura palatina y anquiloglosia. El gen que lo produce inicialmente fue localizado en el cromosoma Xq21, sin embargo posteriormente se vio que alteraciones en el gen Tbx22 también son responsables de la formación de CPX. TBX22 es un factor de transcripción que cumple un importante papel en el desarrollo temprano y en el paso de células epiteliales a mesodérmicas. Adicionalmente mutaciones puntuales determinadas en este gen han sido asociadas con herencia mendeliana ligada al X donde los pacientes además de presentar fisuras labiales y/o palatinas presentan también úvula bífida o ausente y anquiloglosia.36 Investigaciones a nivel molecular han establecido que la expresión del gen Tbx22 está involucrada principalmente en el crecimiento de los procesos palatinos y no en la fusión de ellos. TBX22 es miembro de la familia de factores de transcripción T-box y fue uno de los primeros genes identificados como principal responsable de fisura palatina sindrómica, dado que la alteración de dicho gen en el ratón induce una gran cantidad de anormalidades durante el desarrollo facial.37
En el síndrome velocardiofacial (#192430) se ha observado una deleción en 22q11. Estos pacientes presentan fisuras palatinas, defectos cardíacos, ausencia o hipoplasia del timo, trastornos del habla y problemas auditivos.12
Por otra parte, el síndrome de Treacher Collins (#154500) es un desorden autosómico dominante, que se caracteriza por presentar alteraciones en ojos y oídos, deficiencia malar, pérdida de la audición, micrognatia y coloboma entre otros. Este síndrome se produce por alteraciones en el gen Tcof1 en el cromosoma 5q32-q33.1. Este gen codifica para la proteína treacle importante en el desarrollo embrionario de cara y cabeza. La función exacta de la proteína no se conoce pero se cree que está involucrada en la traslocación de proteínas entre el núcleo y el citoplasma celular.38,39
Las mutaciones en el gen para el péptido señal Shh se han asociado con fenotipo de holoprosencefalia. Mutaciones heterocigotas en familias humanas se han asociado con diversos fenotipos de holoprosencefalia, debido al papel importante de este péptido señal en el desarrollo de las estructuras mediofaciales en el embrión humano.19
Alteraciones en el gen Col2a1 se han relacionado con el síndrome de Stickler (#108300). Este gen codifica para el colágeno tipo II. Uno de los efectos de la alteración en la biosíntesis del colágeno es la morfogénesis esquelética anormal y la inducción de fisura palatina.40
Otros genes vienen siendo estudiados como posibles candidatos de inducción de CL/P. Dentro de ellos se encuentran el receptor del factor de crecimiento de fibroblastos FGFR1. Alteraciones en este gen se han relacionado con el síndrome de Kallman. Sin embargo también se ha encontrado, aunque en menor porcentaje, pacientes con mutaciones en este gen que presentan fisura labial o palatina aislada.30 Mutaciones en el gen que codifica para el factor de transcripción SATB2 y el gen que codifica para la enzima acylcoA desaturasa ACOD4 también se han visto relacionados con formación de fisura palatina y con fisura labial respectivamente.41
Cada uno de los genes mencionados anteriormente cumplen una función primordial durante el proceso de desarrollo embrionario y cualquier alteración en uno o varios de estos genes producirá alteraciones en cualquier fase del proceso de palatogénesis.
Los genes Bmp2, Shh, Tgfb3 han sido asociados con el proceso de eliminación del borde epitelial medial (Figura 2A). En el crecimiento y elevación de los procesos palatinos se ha involucrado principalmente a los genes Msx1, Tbx22, Bmp4, Bmp2, Lhx8 (Figura 2B), y en el proceso de fusión se ha establecido un papel importante para el gen Tgfb3, Irf6 y Pvrl1 entre otros (Figura 2C).41
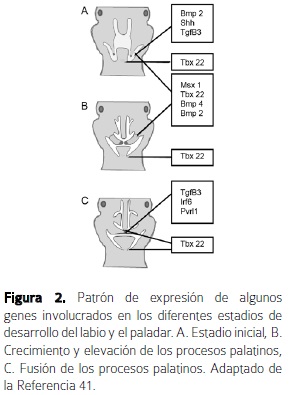
El conocimiento de los genes que participan en el desarrollo craneofacial humano y en la formación de fisuras orofaciales se ha incrementado significativamente en los últimos años. Estos genes proporcionaran herramientas para estudiar y dilucidar las vías genéticas que ellos modulan y cómo ellas se regulan entre sí, permitiendo de este modo en un futuro, el control de estas complejas redes genéticas causantes de este tipo de patologías.
Conclusiones
El estudio y el conocimiento acerca de los mecanismos moleculares que están involucrados en la formación de CL/P viene en aumento gracias al conocimiento del genoma humano en su totalidad y al desarrollo de herramientas modernas de biología molecular que han permitido la identificación de genes candidatos y de las posibles vías de señalización que los mismos están modulando, cuya alteración se ve reflejada en la inducción de estas patologías.
El hallazgo futuro de los genes cuya alteración en la expresión o función esté directamente asociada con la inducción de CL/P contribuirá a las posibilidades de un diagnóstico temprano y la identificación de pacientes altamente susceptibles, así como también al desarrollo de técnicas de modulación genética que permitirán controlar o regular la expresión o función de dichos genes.
Referencias
1. Ferguson M. Palate development. Development. 1988; 103:41-60. [ Links ]
2. Cohen M. Etiology and pathogenesis of orofacial clefting. Oral Maxilofac Surg Clin North Am. 2000;12:379-383. [ Links ]
3. Isaza C, Manrique LA. Anomalías y síndromes asociados con labio y/o paladar hendido. Colomb Med. 1991;20:55-61. [ Links ]
4. Muenke M. The pit, the cleft and the web. Nat Genet. 2002;32:219-220. [ Links ]
5. Dudas M, Kim J, Li WY, Nagy A, Larsson J, Karlsson S, et al. Epithelial and ectomesenchymal role of the type I TGF- receptor ALK5 during facial morphogenesis and palatal fusion. Dev. Biol. 2006;296: 298-314. [ Links ]
6. Dudas M, Li W, Kim J, Yang A, Kaartinen V. Palatal fusion-Where do the midline cells go? A review on cleft palate, amayor human birth defect. Acta Histochem. 2007;109:1-14. [ Links ]
7. Nawshad A, LaGamba D, Hay E. Transforming growth factor b (TGFb) signaling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol. 2004;49:675-689. [ Links ]
8. Jiang R, Bush J and Lidral A. Development of the Upper Lip: Morphogenetic and Molecular Mechanisms Dev Dyn. 2006; 235:1152-1166. [ Links ]
9. Carinci F, Pezzetti F, Scapoli L, Padula E, Baciliero U, Curioni C, et al Non-syndromic clef lip and palate: evidence of linkage to a microsatellite marker on 6p23. Am J Hum Genet. 1995;56: 337-339. [ Links ]
10. Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin -1. Nature 1994, 703-710. [ Links ]
11. Clouthier D, Hosoda K, Richardson J, Williams S, Yanagisawa H, Kuwaki T, et al. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125: 813-824. [ Links ]
12. Cobourne M. The complex genetics of cleft lip and palate. Eur J Orthod. 2004;26:7-16. [ Links ]
13. Carinci F, Pezzetti F, Scapoli L, Martinelli M, Carinci P, Tognon M. Genetics of Nonsyndromic Cleft Lip and Palate: A Review of International Studies and Data Regarding the Italian Population. Cleft Palate Craniofac J. 2000;37:33-40. [ Links ]
14. Prescott N, Winter R, Malcolm S. Non-syndromic clef lip and palate:complex genetics and environmental effects. Ann Hum Genet. 2001;65:5005-515. [ Links ]
15. Dudas M, Nagy A, Laping NJ, Moustakas A, Kaartinen V. Tgf-h3-induced palatal fusion is mediated by Alk-5/Smad pathway. Dev Biol. 2004; 266:96-108. [ Links ]
16. Kohama K, Nonaka K, Hosokawa R, Shum L, Ohishi M. TGFb3 promotes scarless repair of cleft lip in mouse foetuses. J Dent Res. 2002 ;81:688-94. [ Links ]
17. Nawshad A., LaGamba D., Hay E. Transforming growth factor b (TGFb) signaling in palatal growth, apoptosis and epithelial mesenchymal transformation (EMT). Arch Oral Biol. 2004;49:675-689. [ Links ]
18. Van den Boogaard M, Dorland M, Beemer F, van Amstel H. MSX1 mutation is associated with orofacial clefting and tooth agenesis in humans. Nat Genet. 2000; 24:342-343. [ Links ]
19. Gritli-Linde A. Molecular control of secondary palate development. Dev. Biol. 2007;301: 309-326. [ Links ]
20. Zhang Z, Song Y, Zhao X. Rescue of cleft palate in Msx1-deficient mice by transgenic Bmp4 reveals a network of BMP and Shh signaling in the regulation of mammalian palatogenesis. Development. 2002;129:4135-4146. [ Links ]
21. Vieira A, Orioli I, Castilla E, Cooper M, Marazita M, Murray J. MSX1 and TGFB3 Contribute to Clefting in South America. J Dent Res 2003; 82:289-292. [ Links ]
22. Otero L, Gutierrez S, Chaves M, Vargas C, Bermudez L. Association of MSX1 with nonsyndromic cleft lip and palate in a Colombian population. Cleft Palate-Craniofacial J. 2007;44:653-656. [ Links ]
23. Liu W, Sun X, Braut A, Mishina Y, Behringer R, Mina Mina M, et al. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development 2005;132:1453-1461. [ Links ]
24. Lu H, Jin Y, Tipoe G. Alteration in the expression of bone morphogenetic protein-2,3,4,5 mRNA during pathogenesis of cleft palate in balb/c mice. Arch Oral Biol 2000;45:133-40. [ Links ]
25. Nie X. Cranial base in craniofacial development: Developmental features, influence on facial growth, anomaly and molecular basis. Acta Odontol Scand. 2005;63:127-35. [ Links ]
26. Van der Woude A. Fistula labii inferioris congenita and its association with cleft lip and palate. Am J Hum Genet. 1954;6:244-56. [ Links ]
27. Zucchero T, Cooper M, Caprau D, Ribero L, Suzuki Y, Yoshiura K, et al. IRF6 is a major modifier for nonsyndromic cleft with or without cleft palate. Am. J. Hum. Genet. 2003;73, A4. [ Links ]
28. Schutte B, Bjork B, Coppage K, Malik M, Gregory S, Scott D, et al. A preliminary gene map for the van der Woude syndrome critical region derived from 900 kb of genomic sequence at 1q32-q41. Genome Res. 2000;10:81-94. [ Links ]
29. Kondo S, Schutte B, Richardson R, et al. Mutations in IRF6 cause Van der Woude and popliteal pterygium syndromes. Nat Genet. 2002;32:285-9. [ Links ]
30. Prescott N, Garcia-Rodriguez C, Lees M, Winter R. Candidate genes in nonsyndromic cleft lip and palate. Am J Hum Genet. 2003;73: A727. [ Links ]
31. Suzuki K, Hu D, Bustos T, Zlotogora J, Richieri-Costa A, Helms J, et al. Mutations of PVRL1, encoding a cell-cell adhesion molecule/herpesvirus receptor, in cleft lip/palateectodermal dysplasia. Nat Genet. 2000;25:427-30. [ Links ]
32. Cortez-Franco F, Garcia-Salas S, Medina-Florez J. Síndrome de ectrodactilia, displasia ectodérmica y paladar hendido (EEC) y dermatitis: Reporte de un caso. Dermatol. Peru. 2005;15:56-59. [ Links ]
33. Barrow L, van Bokhoven H, Daack-Hirsch S, Andersen T, van Beersum S, Gorlin R, et al. Analysis of the p63 gene in classical EEC syndrome, related syndromes, and non-syndromic orofacial clefts. J. Med Genet. 2002; 39:559-566. [ Links ]
34. Jiang R, Lan Y, Chapman HD, Shawber C, Norton CR, Serrez, DV, et al. Defects in limb, craniofacial, and thymic development in Jagged2 mutant mice. Genes Dev. 1998;12:1046-1057. [ Links ]
35. Cox T, Allen L, Cox L, Hopwood B, Goodwin B, Haan E, et al. New mutations in MID1 provide support for loss of function as the cause of X-linked Opitz syndrome. Hum Mol Genet. 2000;9:2553-62. [ Links ]
36. Braybrook C, Lisgo S, Doudney K, Henderson D, Marcano A, Strachan T, et al. Craniofacial expression of human and murine TBX22 correlates with the cleft palate and ankyloglossia phenotype observed in CPX patients. Hum Mol Genet 2002; 11:2793-2804. [ Links ]
37. Marcano A, Doudney K, Braybrook C, Squires R, Patton M, Lees M, et al. TBX22 mutations are a frequent cause of cleft palate. J Med Genet. 2004; 41:68-74. [ Links ]
38. Wise CA, Chiang L, Paznekas W, Sharma M, Musy M, Ashley J, et al. TCOF1 gene encodes a putative nucleolar phosphoprotein that exhibits mutations in Treacher Collins syndrome throughout its coding region. Proc Natl Acad Sci U S A. 1997;94:3110-5. [ Links ]
39. Andrade E, Júnior V, Didoni A, Freitas P, Carneiro A, Yoshimoto F. Treacher Collins syndrome with choanal atresia: a case report and review of disease features Rev Bras Otorrinolaringol. 2005; 71: 15-18. [ Links ]
40. Snead M, Yates J. Clinical and molecular genetics of stickler syndrome. J Med Genet. 1999;36:353-9. [ Links ]
41. Stanier P, Moore G. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet. 2004;13 Spec No 1:R73-81. [ Links ]
