










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Odontología
Print version ISSN 0120-971X
CES odontol. vol.28 no.1 Medellìn Jan./June 2015
Manifestaciones clínicas del espectro óculo aurículo vertebral
Clinical manifestations of oculo auriculo vertebral spectrum
Sebastián Véliz-Mendez,1 Leonardo González-Escobar,1 Noemí Leiva-Villagra,2 3 Pamela Agurto-Veas,2
1Cirujano Dentista, Unidad de Malformación Craneofacial, Facultad de Odontología. Universidad de Chile. Chile. E-mail: sebastianvelizm@gmail.com
2Ortodoncista. Hospital Luis Calvo Mackenna, Santiago, Chile. E-mail: leivanoemi@yahoo.com
3Profesor asistente, Facultad de Odontología, Directora Unidad de Malformación Craneofacial. Universidad de Chile. Chile. E-mail: pameagurto@yahoo.com
Forma de citar: Véliz-Mendez S, González-Escobar L, Leiva-Villagra Noemí, Agurto-Veas P. Manifestaciones clínicas del espectro óculo aurículo vertebral. Rev. CES Odont 2015; 28(1): 76-85
Recibido: febrero de 2014. Aprobado: junio de 2015
Resumen
El Espectro óculo Aurículo Vertebral corresponde a un desorden heterogéneo y complejo que afecta el desarrollo de estructuras derivadas del primer y segundo arco branquial. Presenta un compromiso variable del macizo maxilofacial, condicionando el tratamiento de cada paciente a su expresión fenotípica. El objetivo de esta revisión de tema es describir las diversas características de este espectro. Para ello se realizó un análisis de la literatura científica con el fin de entregar una referencia actualizada sobre las principales manifestaciones clínicas, su diagnóstico, exámenes complementarios utilizados y posibles diagnósticos diferenciales. Dada su gran variabilidad, es de vital importancia que sea conocido no solo por el odontólogo, sino también por los equipos de salud, posibilitando la entrega de un diagnóstico oportuno a temprana edad y un manejo adecuado de las diferentes alteraciones que pueden presentar.
Palabras clave: Asimetría facial, desarrollo maxilofacial, anomalías craneofaciales.
Abstract
The Oculo Auriculo Vertebral spectrum corresponds to a heterogeneous and complex disorder that affects the development of structures derived from the first and second branchial arch. It has a variable commitment of maxillofacial structures , conditioning treatment for each patient to their phenotypic expression. The objective of this review article it's to describe various features of this spectrum . A scientific literature analysis was done with the aim of delivering an up to date reference regarding the main clinical manifestations, diagnosis, complementary exams used and possible differential diagnosis. Given its variable expression is that it becomes important to be known not only by the dentist, but also by health teams , allowing the delivery of timely early diagnosis and proper management of the different alterations that may present.
Key words: Facial asymmetry, maxillofacial development, craniofacial anomalies
Introducción
El Espectro Oculo-Aurículo-Vertebral (OAVS por sus siglas en inglés) corresponde a una malformación congénita con alteraciones de estructuras desarrolladas a partir del primer y segundo arco branquial. Por ser una anomalía en espectro, sus presentaciones clínicas varían de acuerdo a la severidad de cada paciente, lo que ha llevado también a que existan diversos nombres según las estructuras comprometidas como Microsomía Hemifacial (MHF), Síndrome de Goldenhar, Displasia Oculo-Aurículo-Vertebral, Síndrome del primer arco, Síndrome del primer y segundo arco branquial, entre otros, siendo las dos primeras descritas en forma autónoma. La Microsomía Hemifacial fue definida como una condición que afecta el desarrollo primario mandibular, bucal y auditivo (1); mientras que el Síndrome de Goldenhar fue descrito como una variante, donde se suman alteraciones vertebrales y dermoides epibulbares (2).
Existen diferentes teorías respecto a su etiología, desde los diferentes grados de hemorragia de la arteria estapedial durante el desarrollo embrionario (3), alteración en la migración de las células de la cresta neural (4), alteraciones genéticas autosómicas dominantes (5) o incluso variantes cromosómicas (6).
EpidemiologíaLa incidencia ha sido reportada en un amplio rango, debido a la variabilidad de su expresión, subestimando los casos leves y maldiagnosticando casos más severos en donde hay compromiso de estructuras extracraneales (7). Si bien los reportes van desde 1/3500(8) hasta 1/22550 (9); la estimación más utilizada es la reportada por Grabb de1/5600(10). Se estima que los hombres son más afectados que las mujeres en un rango de 3:2; y que el lado derecho se encuentra comprometido más que el izquierdo en la misma proporción (1).
Manifestaciones ClínicasPor ser una anomalía en espectro, hay una amplia variedad de manifestaciones clínicas, las que en gran medida comprometen estructuras del primer y segundo arco branquial, lo que no implica la ausencia de alteraciones extracraneales. Las principales alteraciones se encuentran a nivel de mandíbula (cuerpo, rama y ATM), órbita, auricular, nervioso y de tejidos blandos.
Órbita y alteraciones ocularesLa Microftalmia (Figura 1A) y la alteración de la posición de la órbita o Distopia Orbitaria (Figura 1B) son las alteraciones más común en estos pacientes. También es frecuente encontrar Blefaroptosis (1), Anoftalmia o Dermoides Epibulbares, ya sean unilateral o bilaterales (11), estos últimos característicos en el Síndrome de Goldenhar. Otros hallazgos frecuentes son coloboma del párpado superior, del iris, coroideo o de retina. (Figura 1B, flecha) (12). Es común que se presenten varias de estas alteraciones a la vez.
Mandíbula y Articulación Témporo Mandibular (ATM)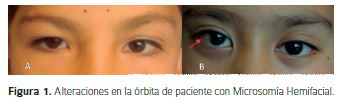
El desarrollo asimétrico por hipoplasia de la mandíbula es una de las principales características descritas, que en su mayoría es unilateral, con un 10% a un 33% de compromiso bilateral (1, 10), presentando un lado más afectado. El grado de compromiso mandibular va desde una leve disminución del tamaño del cuerpo o rama mandibular (Figura 2A) hasta la ausencia de rama, cóndilo, coronoides y cavidad glenoídea del lado afectado (Figura 2B). La ATM puede estar presente pero alterada en ubicación y/o en sus componentes o simplemente puede estar ausente o anquilosada. El disco articular del lado afectado puede presentar alteraciones de acuerdo al individuo, pero no se condice con el grado de hipoplasia mandibular (13). Existe una desviación del mentón y de la línea media dentaria hacia el lado afectado, el cual se mantiene durante la dinámica mandibular.
Nervios Faciales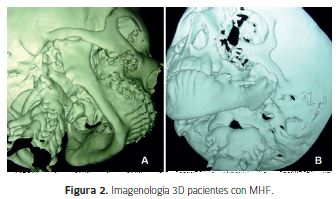
Lo más frecuente de encontrar es el compromiso del Nervio Facial, mientras que en casos más severos puede haber compromiso de los nervios Trigémino e Hipogloso. Esto se manifiesta principalmente a través de parálisis facial en relación a la magnitud del defecto del oído, pero también puede haber Anestesia Trigeminal o elevación asimétrica muscular, desviándose hacia el lado afectado.
Pabellón AuricularEs posible encontrar desde una leve microtia con presencia de todas las estructuras hasta Anotia, con una moderada correlación de la deformidad facial (Figura 3) (14). Hay malposición del pabellón auricular e incluso pérdida de la Audición. También es común encontrar tags preauriculares de piel y cartílago, atresia del meato externo y anomalías del oído medio e interno (12).
Extracraneales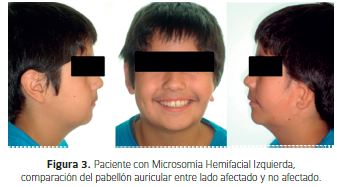
Un estudio realizado por Horgan y Cols. reportó la presencia de anomalías extracraneales en pacientes con MHF, encontrando alteraciones a nivel Renal (Ausencia de Riñón, Doble uréter), Pulmonar, Cardiaco (Defectos del Septo Ventricular, Anomalías del Arco Aórtico), Gastrointestinal (Atresia Duodenal (18)), Esqueletal (Alteraciones Vertebrales como occipitalización del atlas, sinostosis, fusión de vértebras) y en el Sistema Nervioso Central (Malformaciones Cerebrales, Microcefalia, Hidrocefalia) (19).
A su vez, se han reportado otras alteraciones en estos pacientes, como una mayor prevalencia de ronquidos y trastornos del sueño (20); mientras que a nivel psicosocial se ha encontrado una menor aceptación de los pares y competencias sociales pobres (21).
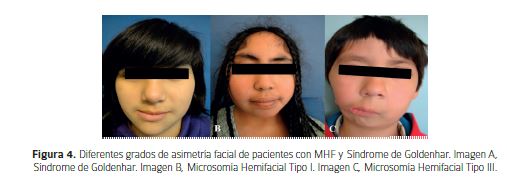
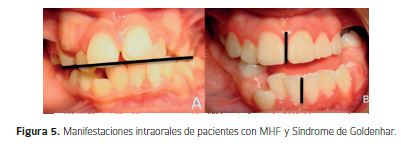
Bucodental y MaxilofacialLa hipoplasia mandibular conlleva a una falta de desarrollo del maxilar, ubicándose ambos en una posición más posterior (22). Presencia de Maloclusiones (Mordida Cruzada en el lado afectado, apiñamiento), agenesias (23), hipoplasias Dentales (24) y retraso en la erupción dentaria (25). Hay una inclinación del plano oclusal hacia arriba en dirección al lado afectado (Figura 5A), menor altura alveolar y desviación de la línea media y mandibular hacia el lado afectado (Figura 5). Se puede observar en un 65% de los pacientes algún grado de asimetría facial, la cual puede no ser evidente en un inicio, pero que se acentúa durante el crecimiento. (1)
Diagnóstico
El diagnóstico de OAVS es clínico, y la participación de un equipo transdisciplinario es esencial debido al amplio rango de alteraciones y la diversidad de anomalías entre cada paciente (26). Por ello, es importante realizar un examen exhaustivo a temprana edad apoyado en exámenes complementarios que permitan la evaluación del compromiso de estructuras como su severidad.
• Radiografía panorámica: Análisis Inicial. Permite una visión general de las estructuras óseas mandibulares y maxilares, así como la relación de las piezas dentarias en el macizo maxilofacial y una evaluación del lado afectado con su contralateral (Figura 6A).
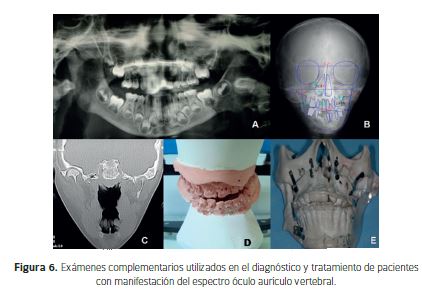
• Telerradiografía de perfil: Permite observar la relación entre el maxilar y la mandíbula, así como de éstos con el cráneo. También se puede analizar el perfil del paciente.
• Radiografía Cefalica Postero-Anterior: Permite observar principalmente las asimetrías y desviación mandibular, sin determinar las estructuras que la causan ni su grado de compensación (Figura 6B).
• Tomografía Computarizada: Permite exploraciones en 3D. Se prefiere en casos más severos, debido a que la fiabilidad de la Radiografía convencional disminuye al aumentar el compromiso de estructuras. (27). Tiene la ventaja de permitir girar las imágenes, obteniendo una visión desde distintos ángulos (28) (Figura 6 C).
• Examen de Modelos: Permite planificar el tratamiento en pacientes con alteración del crecimiento facial, evaluando la oclusión dentaria en los tres sentidos del espacio (Figura 6D).
• Estereolitografía: Indicada en pacientes complejos con el fin de planificar el tratamiento, cirugías y aparatos intraorales para osteodistracción (Figura 6E)
• Ultrasonido: Durante el embarazo se pueden observar a través del ultrasonido 2D y 3D alteraciones como tags preauriculares, microftalmia y fisura palatina, las que en su conjunto permite acercarnos a un diagnóstico temprano (29).
Diagnóstico Diferencial
Debido a que el Espectro óculo Aurículo Vertebral es altamente variable fenotípicamente y causalmente heterogéneo; es necesario realizar un diagnóstico diferencial con otros síndromes, siendo los principales a contrastar:
• Síndrome Treacher Collins: Corresponde a un desorden del desarrollo facial de origen autosómico dominante, el cual se origina por defectos en la formación de las células de la cresta neural. (30) Las principales características de este síndrome se presentan de manera bilateral simétrica y son displasia o ausencia de la región órbito malar, defectos en la articulación temporomandibular (ATM) y/o ramas mandibulares y cóndilos hipoplásicos, fisuras palpebrales con inclinación antimongoloide, retromicrognatia con mordida abierta anterior severa y maloclusión clase II esqueletal, nariz desproporcionadamente grande y en forma de pico, hipoplasia maxilar anormal o ausencia de los músculos de la masticación (particularmente pterigoideos y maseteros), osículo defectuosos, hipoacusia conductiva, coloboma oftálmica (31), alteraciones en el tamaño, forma, y posición de las orejas externas, posibles problemas del desarrollo del cerebro como microcefalia y deficiencia intelectual y psicomotora (32).
• Disostosis acrofacial preaxial o Síndrome de Nager: Pertenece a un desorden del primer y segundo arco branquial, con causa desconocida, se han postulado la acción de distintos genes tanto de herencia recesiva como dominante. Las características típicas de este síndrome incluyen fisuras palpebrales inclinadas hacia abajo, aplanamiento malar, malformaciones del oído externo, falta de musculatura periorbicular impidiendo el cierre bucal, anomalías en la mano como pequeños pulgares y, en algunos casos, defectos de reducción de extremidades superiores (33-35).
• Síndrome de Miller o Disostosis acrofacial postaxial: Las anomalías craneofaciales son similares a los pacientes con Treacher Collins. Los defectos en las extremidades predominantemente implican el rayo postaxial, con una amplia gama de gravedad, y en menor medida el rayo preaxial. Las principales características que presenta son hipoplasia malar, micrognatia, colobomas, aplasia medial de las pestañas del párpado inferior, paladar con fisura, orejas ahuecadas y deficiencia de rayo cubital (36).
• Síndrome de CHARGE: Su nombre corresponde a una nemotecnia en inglés CHARGE (Coloboma, Heart defects, choanal Atresia, Retarded growth and development, Genital abnormalities, and Ear anomalies), la cual es de origen heterogéneo. La gran mayoría de las características de la enfermedad se muestran en el período prenatal, mientras que algunos problemas debido a la carga como retraso en el crecimiento o retraso pueden ser adquiridas en el período postnatal como resultado de la enfermedad. Las principales características del síndrome son coloboma ocular, atresia o estenosis de coanas, disfunción del nervio craneal y anomalías del oído característico. Otros signos en menor medida son facies CHARGE característica, paladar fisurado, malformaciones cardiovasculares, fístula traqueoesofágica, hipoplasia genital, retraso en el desarrollo y deficiencia de crecimiento (37, 38).
• Síndrome de Townes-Brocks: Síndrome de baja incidencia, con transmisión hereditaria autosómica dominante. Los principales signos que caracterizan este síndrome, los cuales pueden estar en mayor o menor medida, corresponden a anomalías de las orejas con sordera neurosensorial o hipoacusia, polidactilia preaxial (radial), pulgar trifalángico u otra anomalía del pulgar, ano imperforado o malposición anal y malformaciones renourinarias (39, 40).
Conclusión
La variable expresión de características observadas en pacientes con Espectro óculo Aurículo Vertebral dificulta notablemente su diagnóstico, especialmente en casos leves y severos. Esto explica tan amplio rango de incidencia a nivel mundial, ya que en Chile no existe una incidencia estimada. Su expresión en el macizo maxilofacial no sólo es resorte de la odontología, sino que también de distintas áreas de la salud, por lo que el conocimiento por parte de los equipos de salud puede ayudar a una pesquisa temprana y un tratamiento oportuno.
Referencias
1. Gorlin RJ CMJ, Hennekam RCM. . Syndromes of the Head and Neck, 4th edition. New York: Oxford University Press; 2001. p. 790. [ Links ]
2. Gorlin RJ, Jue KL, Jacobsen U, Goldschmidt E. Oculoauriculovertebral Dysplasia. Pediatrics. 1963;63:991-9. [ Links ]
3. Poswillo D. The pathogenesis of the first and second branchial arch syndrome. Oral Surg Oral Med Oral Pathol. 1973;35(3):302-28. [ Links ]
4. Johnston MC, Bronsky PT. Animal models for human craniofacial malformations. J Craniofac Genet Dev Biol. 1991;11(4):277-91. [ Links ]
5. Tasse C, Majewski F, Bohringer S, Fischer S, Ludecke HJ, Gillessen-Kaesbach G, et al. A family with autosomal dominant oculo-auriculo-vertebral spectrum. Clin Dysmorphol. 2007;16(1):1-7. [ Links ]
6. Choong YF, Watts P, Little E, Beck L. Goldenhar and cri-du-chat syndromes: a contiguous gene deletion syndrome? J AAPOS. 2003;7(3):226-7. [ Links ]
7. Hartsfield JK. Review of the etiologic heterogeneity of the oculo-auriculo-vertebral spectrum (Hemifacial Microsomia). Orthod Craniofac Res. 2007;10(3):121-8. [ Links ]
8. Poswillo D. Otomandibular deformity: pathogenesis as a guide to reconstruction. J Maxillofac Surg. 1974;2(2-3):64-72. [ Links ]
9. Melnick M. The etiology of external ear malformations and its relation to abnormalities of the middle ear, inner ear, and other organ systems. Birth defects Orig Artic Ser. 1980;16(4):303-31. [ Links ]
10. Grabb WC. The first and second branchial arch syndrome. Plast Reconstr Surg. 1965;36(5):485-508. [ Links ]
11. Baum JL, Feingold M. Ocular aspects of Goldenhar's syndrome. Am J Ophthalmol. 1973;75(2):250-7. [ Links ]
12. Kokavec R. Goldenhar syndrome with various clinical manifestations. Cleft Palate Craniofac J. 2006;43(5):628-34. [ Links ]
13. Kitai N, Murakami S, Takashima M, Furukawa S, Kreiborg S, Takada K. Evaluation of temporomandibular joint in patients with hemifacial microsomia. Cleft Palate Craniofac J. 2004;41(2):157-62. [ Links ]
14. Figueroa AA, Pruzansky S. The external ear, mandible and other components of hemifacial microsomia. J Maxillofac Surg. 1982;10(4):200-11. [ Links ]
15. Fan WS, Mulliken JB, Padwa BL. An association between hemifacial microsomia and facial clefting. J Oral Maxillofac Surg. 2005;63(3):330-4. [ Links ]
16. Heude E, Rivals I, Couly G, Levi G. Masticatory muscle defects in hemifacial microsomia: a new embryological concept. Am J Med Genet A.2011;155A(8):1991-5. [ Links ]
17. Funayama E, Igawa HH, Nishizawa N, Oyama A, Yamamoto Y. Velopharyngeal insufficiency in hemifacial microsomia: analysis of correlated factors. Otolaryngol Head Neck Surg. 2007;136(1):33-7. [ Links ]
18. Hacihamdioglu B, Unay B, Hacihamdioglu DO, Vurucu S, Gul D, Akin R. Goldenhar syndrome with duodenal atresia: a new finding. Clin Dysmorphol. 2008;17(2):141-2. [ Links ]
19. Horgan JE, Padwa BL, LaBrie RA, Mulliken JB. OMENS-Plus: analysis of craniofacial and extracraniofacial anomalies in hemifacial microsomia. Cleft Palate Craniofac J. 1995;32(5):405-12. [ Links ]
20. Cloonan YK, Kifle Y, Davis S, Speltz ML, Werler MM, Starr JR. Sleep outcomes in children with hemifacial microsomia and controls: a follow-up study. Pediatrics. 2009;124(2):e313-21. [ Links ]
21. Dufton LM, Speltz ML, Kelly JP, Leroux B, Collett BR, Werler MM. Psychosocial outcomes in children with hemifacial microsomia. J Pediatr Psychol. 2011;36(7):794-805. [ Links ]
22. Ongkosuwito EM, van Neck JW, Wattel E, van Adrichem LN, Kuijpers-Jagtman AM. Craniofacial morphology in unilateral hemifacial microsomia. Br J Oral Maxillofac Surg. 2013 Dec;51(8):902-7. [ Links ]
23. Maruko E, Hayes C, Evans CA, Padwa B, Mulliken JB. Hypodontia in hemifacial microsomia. Cleft Palate Craniofac J. 2001;38(1):15-9. [ Links ]
24. Farias M, Vargervik K. Tooth size and morphology in hemifacial microsomia. Int J Paediatr Dent. 1998;8(3):197-201. [ Links ]
25. Ongkosuwito EM, de Gijt P, Wattel E, Carels CE, Kuijpers-Jagtman AM. Dental development in hemifacial microsomia. J Dent Res. 2010;89(12):1368-72. [ Links ]
26. Association AC-P-C. Parameters for the Evaluation and Treatment of Patients with Cleft Lip/Palate or Other Craniofacial Anomalies. Revised Edition 2009. Original Publication Cleft Palate-Craniofacial Journal 1993;30 (Suppl 1). [ Links ]
27. Takahashi-Ichikawa N, Susami T, Nagahama K, Ohkubo K, Okayasu M, Uchino N, et al. Evaluation of mandibular hypoplasia in patients with hemifacial microsomia: a comparison between panoramic radiography and three-dimensional computed tomography. Cleft Palate Craniofac J. 2013;50(4): 381-7. [ Links ]
28. Birgfeld CB, Saltzman BS, Luquetti DV, Latham K, Starr JR, Heike CL. Comparison of two-dimensional and three-dimensional images for phenotypic assessment of craniofacial microsomia. Cleft Palate Craniofac J. 2013;50(3):305-14. [ Links ]
29. Volpe P, Gentile M. Three-dimensional diagnosis of Goldenhar syndrome. Ultrasound Obstet Gynecol. 2004;24(7):798-800. [ Links ]
30. Trainor PA, Andrews BT. Facial dysostoses: Etiology, pathogenesis and management. Am J Med Genet C Semin Med Genet. 2013;163(4):283-94. [ Links ]
31. Green B, Nikkhah D, Cobb AR, Dunaway DJ. Craniofacial disorders that have phenotypic overlap with Treacher Collins syndrome: J Plast Reconstr Aesthet Surg. 2013 Aug;66(8):e234-5. doi: 10.1016/j. bjps.2013.04.029. [ Links ]
32. Dixon J, Trainor P, Dixon MJ. Treacher Collins syndrome. Orthod Craniofac Res. 2007;10(2):88-95. [ Links ]
33. Bernier FP, Caluseriu O, Ng S, Schwartzentruber J, Buckingham KJ, Innes AM, et al. Haploinsufficiency of SF3B4, a component of the pre-mRNA spliceosomal complex, causes Nager syndrome. Am J Hum Genet. 2012;90(5):925-33. [ Links ]
34. Heike CL HA. Craniofacial Microsomia Overview. In: Pagon RA, Adam MP, Bird TD, et al, editors GeneReviews [Internet]Seattle (WA): University of Washington, Seattle; 1993-2013. Disponible en: http://wwwncbinlmnihgov/books/NBK5199/. 2009 [ Links ]
35. Kavadia S, Kaklamanos EG, Antoniades K, Lafazanis V, Tramma D. Nager syndrome (preaxial acrofacial dysostosis): a case report. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2004;97(6):732-8. [ Links ]
36. Ogilvy-Stuart AL, Parsons AC. Miller syndrome (postaxial acrofacial dysostosis): further evidence for autosomal recessive inheritance and expansion of the phenotype. J Med Genet. 1991;28(10): 695-700. [ Links ]
37. Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, et al. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43(3):211-7. [ Links ]
38. Hsueh KF, Yang CS, Lu JH, Hsu WM. Clinical characteristics of CHARGE syndrome. J Chin Med Assoc. 2004;67(10):542-6. [ Links ]
39. Rodriguez Asensio J, Rodriguez Rosell MV, Ramos Perez A. Hearing loss in Townes-Brocks syndrome. Acta Otorrinolaringol Esp. 2003;54(7):518-22. [ Links ]
40. Harrison SJ, Nishinakamura R, Jones KR, Monaghan AP. Sall1 regulates cortical neurogenesis and laminar fate specification in mice: implications for neural abnormalities in Townes-Brocks syndrome. Dis Model Mech. 2012;5(3):351-65. [ Links ]
