










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkCES Odontología
Print version ISSN 0120-971X
CES odontol. vol.30 no.1 Medellìn Jan./June 2017
Artículo de revisión
Cuidado odontológico de pacientes con trastornos hereditarios de la coagulación
Dental care for coagulation hereditary disorder patients
Meliza Andrea Cano-Franco1, Gustavo Eduardo Ortiz-Orrego2, Sandra Elizabeth González- Ariza3
1 Odontóloga Universidad CES. melizacano@gmail.com
2Cirujano Maxilofacial Universidad CES. Master en Tratamiento de dolor Universidad de Salamanca, Docente pregrado y postgrado Universidad CES. gortizo@hotmail.com
3Odontóloga, Magister en Epidemiología, Docente facultad de odontología Universidad CES. sgonzales@ces.edu.co
Forma de citar: Cano-Franco MA, Ortiz-Orrego GE, González-Ariza SE. Cuidado odontológico de pacientes con trastornos hereditarios de la coagulación Rev. CES Odont 2017; 30(1): 30-40.
Recibido: junio de 2016. Aceptado: junio de 2017
Resumen
Los trastornos hereditarios de la coagulación como la hemofilia que es una enfermedad genética ligada al cromosoma X que se manifiesta por la deficiencia de los factores de la coagulación VIII, IX y XI -Hemofilia A, B y C respectivamente. , de acuerdo a la cantidad de deficiencia de estos se clasifican en leve, moderada o severa. Otra de enfermedad que se ve relacionada al trastorno de la coagulación es conocida como enfermedad de Von Willebrand cuya proteína con el mismo nombre se encuentra ausente o disminuido, otro aspecto a resaltar es que esta enfermedad no se encuentra ligada al sexo.
El correcto diagnóstico clínico y exámenes de laboratorio, hace parte de un número de pasos que debe tener en cuenta el odontólogo para realizar normas de atención adecuadas según el tratamiento de cada paciente, ya sea consulta programada para realizar procedimientos como: operatoria, endodoncia, periodoncia, exodoncias, procedimientos de cirugía oral; O que se deba realizar tratamientos de urgencias como: heridas de la mucosa, laceraciones en boca, trauma facial, abscesos o celulitis, trauma dentoalveolar, enfocándose no solo en la parte clínica sino en el adecuado manejo del dolor de cada paciente.
Palabras clave: Trastornos hemorrágicos, hemofia, enfermedad de Von Willebrand.
Abstract
Hereditary disorders of coagulation are genetic disease, among them are hemophilia that is attached to the X chromosome, so it manifests itself in men; In them there is deficiency of coagulation factors VIII, IX and XI -Hemofilia A, B and C respectively-, according to the amount of deficiency of these are classified as mild, moderate or severe. Another disease that is related to the coagulation disorder is known as Von Willebrand disease whose protein with the same name is absent or diminished, another aspect to emphasize is that this disease is not linked to sex.
The correct clinical diagnosis and laboratory tests are part of a number of steps that the dentist must take into account to make adequate standards of care according to the treatment of each patient, whether scheduled consultation to perform procedures such as: surgery, endodontics, periodontics , dental extractions, oral surgery procedures; Or that it is necessary to carry out emergency treatments such as: mucosal wounds, lacerations in the mouth, facial trauma, abscesses or cellulitis, dentoalveolar trauma, focusing not only on the clinical part but also on the adequate management of the pain of each patient.
Keywords: Hemorrhagic disorders, hemophilia, von Willebrand disease.
Introducción
Los trastornos hemorrágicos son un grupo de afecciones en las cuales hay un problema con el proceso de coagulación sanguínea del cuerpo, estos trastornos pueden llevar a que se presente un sangrado intenso y prolongado después de una lesión, el sangrado también puede producirse de manera espontánea afectando internamente tejidos y órganos (1-3).La promoción y prevención en salud oral para estos pacientes hace parte del nivel de atención primaria, es fundamental tener un buen trato con el paciente, guiarlo para tener un buen plan de tratamiento, que el paciente tenga conocimiento de la importancia de realizar interconsulta con su odontólogo(2). Desde finales del segundo siglo antes de Cristo se han reportado casos de enfermedades genéticas derivadas de un defecto recesivo de un cromosoma X, generándose de este modo un déficit en la actividad de los factores VIII, IX y XI de la coagulación y otras discrasias que comprometen la hemostasia como el déficit de factor de Von Willebrand (4). Adicionalmente al requerir estos factores en algunas situaciones el paciente corre un riesgo importante de contagiarse con enfermedades de trasmisión sistémica como el VIH entre otras (5).
El objetivo de esta revisión es darle a los lectores una guia para la atención odontólogica de pacientes con trastornos de la coagulación, tanto hemofilicos como los que sufre enfermedad de Von Willebrand. Se hizo una búsqueda de literatura para obtener los resultados de cómo realizar un adecuado y oportuno tratamiento en estos pacientes.
Estrategia de búsqueda
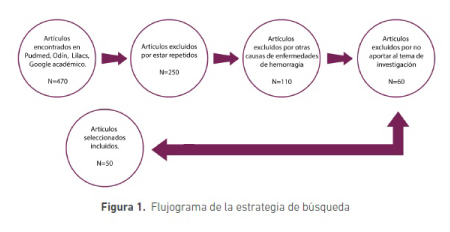
Se realizó una búsqueda de artículos relacionados con salud oral en pacientes con trastornos de la hemorragia y otras discrasias en odontología, las bases de datos fueron Pud Med, Odín, Lilacs, Google Académico. Las palabras clave utilizadas fueron, trastornos hemorrágicos, cuidados en pacientes con enfermedad sistémica, manejo del dolor, salud oral. La cantidad y los criterios de selección de los artículos se muestran en la figura 1.
Hemofilia
La hemofilia es una enfermedad genética derivada de un defecto recesivo en un gen del cromosoma X(1), generándose de este modo un déficit en la actividad de los factores VIII, IX y XI de la coagulación y otras discrasias que comprometen la hemostasia, como consecuencia de lo cual pueden producirse:
- Hemorragia espontánea
- Hemorragia por traumas leves
- Hemorragias producidas por intervenciones odontológicas quirúrgicas
- Hemorragias producidas por intervenciones médico-quirúrgica
Para el hemofílico la mayoría de episodios hemorrágicos se presentan internamente, comprometiendo las mucosas, músculos, articulaciones y el sistema nervioso central, ocasionando daños y secuelas que pueden conducir incluso hasta la muerte (6,7).
Adicionalmente, al requerir en algunas situaciones, reemplazo del factor requerido por medio de hemoderivados, el paciente corre un riesgo importante de contagiarse con enfermedades de transmisión sistémica, tipo hepatitis, VIH, entre otras. Es importante para el entendimiento de la enfermedad, comprender como se da el proceso de la coagulación, proceso en el cual participan una serie de componentes vasculares y sanguíneos para controlar cualquier pérdida sanguínea (8).
Mecanismo por el cual ocurre la coagulación
La hemostasia es un mecanismo de defensa que promueve la integridad de los vasos sanguíneos y evita la pérdida sanguínea. Al ocurrir un daño a nivel de los vasos (ruptura de la capa endotelial) se exponen las proteínas sanguíneas a la capa subendotelial y genera la activación de tres mecanismos, explicados a continuación: (9,10).
1. Fase vascular: reflejo vasoconstrictor reduciendo flujo sanguíneo del vaso afectado.
2. Fase plaquetaria: se forma el taponamiento plaquetario, se adhieren las plaquetas a las fibras de colágeno expuestas de la capa vascular dañada, uniéndose unas con otras.
3. Fase plasmática: hay producción de fibrina la que refuerza el tapón plaquetario. El proceso incluye la transformación de fibrinógeno (soluble) en fibrina (insoluble), por la vía del metabolismo de la trombina, producto de la protrombina. La transformación de la protrombina en trombina toma lugar en dos vías, la intrínseca y la extrínseca (7). La vía intrínseca es activada por el factor de la coagulación XII, como resultado de su contacto con la capa subendotelial dañada. La extrínseca se activa cuando la sangre entra en contacto con la tromboplastina liberada de los tejidos dañados, resultando en la activación del factor VII.
Clasificación
La hemofilia se puede clasificar de acuerdo al factor de la coagulación deficiente en A, B o C y de acuerdo a la concentración del mismo en leve, moderada y severa (11,12).
Hemofilia A
En la hemofilia A, también llamada Hemofilia Clásica, existe deficiencia del factor VIII de la coagulación, que se transmite con carácter recesivo ligado al cromosoma X, en estos casos la mujer es portadora por contar con dos cromosomas X, pero es el hombre quien presenta las manifestaciones de la enfermedad, ya que solo presenta un cromosoma X (figura 2).
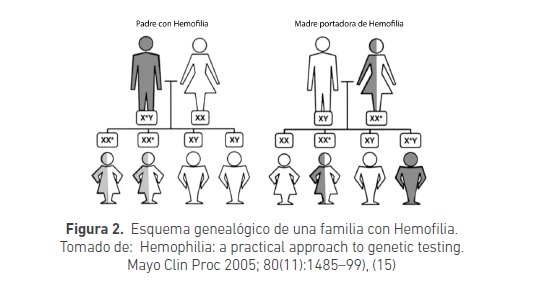
La frecuencia en la población general es de 1/5000 niños nacidos vivos, las mujeres son susceptibles a ser portadoras pero no la sufren. La única excepción es en el raro caso que el padre sea hemofílico y la madre portadora (13). Para el tratamiento se debe tener en cuenta que la vida media del factor son 12 horas, además, debe saberse que el 30 % de los pacientes presentan inhibidores del factor, lo que dificulta más el tratamiento (14).
Hemofilia B
En la hemofilia B, que es también llamada enfermedad de Christmas es una forma de hemofilia caracterizada por la deficiencia del factor IX de la coagulación, que se transmite con carácter recesivo ligado al cromosoma X que afecta principalmente a varones por tener solo una copia del gen X, mientras que las mujeres tienen dos copias del X de modo que si el gen del factor IX en uno de los cromosomas es defectuoso, el gen en el otro cromosoma puede producir suficiente factor IX, aunque este tipo de hemofilia es 5 veces menos común que la hemofilia A, de acuerdo con la concentración del factor, la enfermedad se clasifica así: leve (5 a 50 % de nivel de actividad del factor), moderada (1 a 5 % de nivel de actividad del factor) y severa (menos del 1 % de nivel de actividad del factor) (15).
Las manifestaciones clínicas son clínicamente indistinguibles de la hemofilia A, el diagnóstico se basa en un tiempo prolongado de TPT, con TP y tiempo de sangría normal, el tratamiento es semejante al de la hemofilia A, para el tratamiento se debe tener en cuenta que la vida media del factor son 24 horas (16).
Hemofilia C
En la hemofilia C existe deficiencia del factor XI de la coagulación, que se transmite con carácter recesivo ligado al cromosoma X, Igual que las anteriores de acuerdo a la concentración del factor, la enfermedad se clasifica así: leve (5 a 50 % de nivel de actividad del factor), moderada (1 a 5 % de nivel de actividad del factor) y severa (menos del 1 % de nivel de actividad del factor) (17).
Enfermedad de Von Willebrand
Es la enfermedad adquirida hemorrágica más común en los humanos, afecta el 1-2 % de la población en general, se trata de una enfermedad hereditaria, no ligada al sexo, que produce disminución o ausencia de la proteína sanguínea factor de Von Willebrand, encargada de promover la adhesión plaquetaria a la pared del vaso y de su posterior agregación, también se encarga de transportar el factor VIII, y al faltar dicha proteína el factor es rápidamente degradado, provocando deficiencia, se caracteriza por un prolongado tiempo de sangría y bajos títulos de factor VIII. En estadios leves y moderados se diferencia de las hemofilias en que presenta equimosis y petequias superficiales, lo mismo que hemorragias en membranas mucosas, además, no presenta hemorragias articulares profundas, musculares ni hematomas, como sí ocurre en las hemofilias (18).
Diagnostico de la Hemofilia y Enfermedad de Von Willebrand
Para un adecuado diagnóstico de las enfermedades de la coagulación se requieren:
1. Anamnesis: donde se hará énfasis en antecedentes familiares sobre trastornos hemorrágicos, historia personal de hemorragias (forma como ocurrieron, procedimientos para controlarlas, tiempo de duración y consecuencias) y un completo examen físico.
2. Exámenes de laboratorio: los siguientes son prioritarios: tiempo de sangría, recuento plaquetario, retracción del coagulo, tiempo de trombina (PT), actividad del factor VIII, actividad del factor IX, ensayos de fibrinógeno, pruebas de inhibidores. Para la enfermedad de Von Willebrand (vWF.Ag); actividad de Von Willebrand, análisis de los multímetros de Von Willebrand, agregabilidad plaquetaria con bajas concentraciones de ristocetina. Para todos los pacientes se deben solicitar exámenes de hepatitis B y C, Chagas, sífilis y VIH, ya que en estos casos los pacientes requieren de suministro plasmáticos (19).
Tratamiento Las enfermedades hemorrágicas tales como hemofilia y enfermedad de von willebrand, conlleva, a que los pacientes y sus familiares desarrollen una serie de conductas y comportamientos específicos. El tratamiento integral comprende los aspectos médicos sociales, económicos, de rehabilitación y psicológicos, que permitan al paciente ser consecuente consigo mismo, con los demás y con la sociedad, dentro del tratamiento del paciente y de sus familias se debe incluir el tratamiento psicológico con el fin de ayudar a entender y manejar los efectos de su enfermedad, mejorando para ellos su calidad de vida y su funcionamiento biológico mediante charlas educativas. En cuanto al cuidado oral en odontología, lo más importante es tomar medidas de prevención de la enfermedad como enseñanza cepillado dental, de uso de seda dental y de enjuagues con fluoruros, así mismo, control de la dieta cariogénica y visitas a consulta odontológica frecuente (20-23).
Se debe tener en cuenta la importancia en el tratamiento odontológico, este hace parte de un tratamiento general del paciente y debe ser considerado dentro del manejo integral, debe ser multidisciplinario el abordaje de estos pacientes, lo que implica que cuando sea atendido por odontología debe ser evaluado por hematólogo para llevar a un feliz término el tratamiento. En cuanto al tratamiento odontológico puede realizarse en dos escenarios diferentes: consulta programada y consulta no programada (urgencias) (24,25) (figura 3,4). De manera posterior a las intervenciones en pacientes con trastornos de la coagulación, y como tratamiento alternativo a las lesiones que puede presentar el paciente, tanto en urgencias como en consulta programada, se puede realizar: enjuagues con preparado de agua destilada 200cc con una ampolla de ácido tranexamico durante el primer día, y agua destilada 500cc con ampolla de ácido tranexamico durante los días siguientes, si continúa el sangrado. Poner hielo de manera intermitente por 10 minutos (43-46).
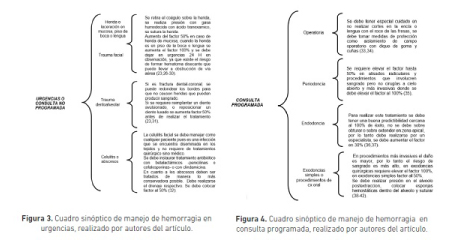
Manejo del dolor
Se debe utilizar medicamentos que no actúen alterando la función plaquetaria -antiagregación plaquetaria-; lo que excluye a los AINES y al ASA, por lo cual se deben utilizar el paracetamol o acetaminofén como medicamento de elección o se pueden utilizar opiáceos.
- Acetaminofén 500 mg a 1 g, cada cuatro a seis horas, durante cuatro a cinco días.
- Acetaminofén + Codeína o Tramadol, ampollas de 50 - 100 mg, tabletas de 50 mg, o gotas de 2.5 mg, cada cuatro a seis horas, sin sobrepasar los 400 mg/día, por cuatro a cinco días, otros métodos para controlar el dolor son antihistamínicos, anestésicos locales y los medios físicos como hielo y calor, ultrasonidos y terapias alternativas entre otros (47-49).
Avances en el manejo de la Hemofilia y Enfermedad de Von Willebrand
Los últimos avances en la terapia genética aplicados a la hemofilia son con el fin de corregir el defecto molecular en el gen mutado, adicionando genes normales que codifican el factor VIII, o el IX en el caso de hemofilia B, basados en tecnología recombinante; este tipo de terapia genética, ofrece la posibilidad de una verdadera sanación, pero por el momento solo se han realizado dichas investigaciones en animales y todavía no puede ser aplicado a los humanos.
En la actualidad, los diferentes manejos que se les brindan a pacientes con hemofilias severas han cambiado sus perspectivas, pudiendo tener una vida con pocas restricciones. Aun no se tiene evidencia suficiente de un avance significativo para tratar la enfermedad de Von Willebrand (50).
Conclusión
El tratamiento incorrecto o insuficiente por parte de odontólogos, en pacientes con trastornos de la coagulación puede llevar al paciente a sufrir consecuencias graves para su salud, debido a esto es importante que los profesionales de la salud tengan un enfoque de tratamiento temprano y adecuado también tener un conocimiento respectivo de cada factor de coagulación el cual puede evitar complicaciones que conlleva a tener pronósticos más favorables y tratamientos más exitosos.
Bibliografía
1. Gupta A. Epstein JB. Cabay RJ. Bleeding disorders of importance in dental care and related patient managment. JCDA. 2007:73;1:77-83a. [ Links ]
2. Israel S. Scwetz N. Boyar R. McNicol A. Bleeding Disorders: Characterization, dental considerations and managment. J Can dent Assoc. 2006;72(9):827-827I. [ Links ]
3. Nagar LK. Sharma N. Khinchi MP. Khan MS. Kumar A. Haemophilia: an overwiew. Asian J Pharmaceut Res Develop. 2017;5(2):1.10. [ Links ]
4. Giannelli F, Green PM, Naylor JA. A genetic view on the etiology of the inhibitor complication [letter]. Blood. 1996;87(6):2612-2612. [ Links ]
5. Ingram GI. The history of haemophilia. J Clin Pathol. junio de 1976;29(6):469-479. [ Links ]
6. Darby SC, Kan SW, Spooner RJ, Giangrande PLF, Hill FGH, Hay CRM, et al. Mortality rates, life expectancy, and causes of death in people with hemophilia A or B in the United Kingdom who were not infected with HIV. Blood. 2007;110(3):815-825. [ Links ]
7. Coppola A, Di Capua M, De Simone C. Primary prophylaxis in children with haemophilia. Blood Transfus.2008;6(2):4-11. [ Links ]
8. Fundaciòn de la Hemofilia. Guía para el manejo de la Hemofilia Congénita [Internet]. 2.a ed. Buenos Aires; 2015. Recuperado a partir de: [ Links ]
9. Jover-Cerveró A, Poveda-Roda R, Bagán JV, Jiménez-Soriano Y. Dental treatment of patients with coagulation factor alterations: An update. Medicina Oral, Patología Oral y Cirugía Bucal (Internet). 2007;12(5):380-387. [ Links ]
10. Stachnik J. Hemophilia: Etiology, complications, and current options in management [Internet]. 2010 July [citado 4 de febrero de 2016] [ Links ].
11. Roberts HR, Monroe DM, White GC. The use of recombinant factor VIIa in the treatment of bleeding disorders. Blood.2004;104(13):3858-3864. [ Links ]
12. Butenas S, Mann KG. Blood coagulation. Biochemistry Mosc. enero de 2002;67(1):3- 12. [ Links ]
13. Genetics: Linkage Analysis [Internet]. [citado 4 de febrero de 2016]. Disponible en: [ Links ]
14. Huth-Kuhne A, Baudo F, Collins P, Ingerslev J, Kessler CM, Levesque H, et al. International recommendations on the diagnosis and treatment of patients with acquired hemophilia A. Haematologica.2009;94(4):566-575. [ Links ]
15. Rossbach H-C. Review of antihemophilic factor injection for the routine prophylaxis of bleeding episodes and risk of joint damage in severe hemophilia A. Vasc Health Risk Manag. 2010;6:59-68. [ Links ]
16. Rodriguez NI, Hoots WK. Advances in Hemophilia: Experimental Aspects and Therapy. Hematology/oncology clinics of North America. 2010;24(1):181-198. [ Links ]
17. Martlew VJ. Peri-operative management of patients with coagulation disorders. Br J Anaesth.2000;85(3):446-455. [ Links ]
18. Saba HI, Tran DQ. Challenges and successes in the treatment of hemophilia: the story of a patient with severe hemophilia A and high-titer inhibitors. J Blood Med. 2012;3:17-23. [ Links ]
19. Wildgoose P, Nemerson Y, Hansen LL, Nielsen FE, Glazer S, Hedner U. Measurement of basal levels of factor VIIa in hemophilia A and B patients. Blood. 1992;80(1):25-28. [ Links ]
20. Kakar, P N, Nishkarsh Gupta, Pradeep Govil, y Vikram Shah.Efficacy and Safety of Tranexamic Acid in Control of Bleeding Following TKR: A Randomized Clinical Trial. Indian J Anaesth.2009;53(6):667-671. [ Links ]
21. Rayen R. Hariharan VS. Elavazhagan N. Kamalendran N. Varadajan R. Dental managment of hemophilic child under general anesthesia. J Indian Soc Prev Dent. 2011;29:74-79. [ Links ]
22. Kalsi H. Nanayakkara L. Pasi KJ. Bowlws L. Hart DP. Access to primary dental care for patients with inherited bleeding disorders. Haemophilia:2012;18:510-515. [ Links ]
23. Tamagond SB. Hugar SI. Patil A. Huddar SR. Chrismas disease: diagnosis and managment of a haemorrhagic diathesis following dentofacial trauma. BMJ. Case Rep 2015. Published online: doi:10.1136/bcr-2014-203790. [ Links ]
24. Rayen R, Hariharan VS, Elavazhagan N, Kamalendran N, Varadarajan R. Dental management of hemophiliac child under general anesthesia. Journal of Indian Society of Pedodontics and Preventive Dentistry.2011;29(1):74-79. [ Links ]
25. Stubbs M. Lloyd J. A protocol for the dental managment of von Willebrand's disease, haemophilia A and haemophilia B. Australian Dent J. 2001;46(1):37-40. [ Links ]
26. Tagliaferri A, Di Perna C, Rivolta GF. Secondary prophylaxis in adolescent and adult haemophiliacs. Blood Transfus.2008;6(2):17-20. [ Links ]
27. Press D. The management of hemophilia in elderly patients. Clinical interventions in aging.2007;2(3):361-368. [ Links ]
28. Madhok R, York J, Sturrock RD. Haemophilic arthritis. Ann Rheum Dis.1991;50(8):588-591.
[ Links ]
29. Rodríguez-Merchán EC. Management of the orthopaedic complications of haemophilia. J Bone Joint Surg Br. marzo de 1998;80(2):191-6. [ Links ]
30. Furie B, Limentani SA, Rosenfield CG. A Practical Guide to the Evaluation and Treatment of Hemophilia. Blood.1994;84(1):3-9. [ Links ]
31. Zafra JMR, Nieto JAC. Guía de actuación en pacientes odontológicos anticoagulados en Atención Primaria. The J Am Dent Assoc. 2010;5(6):272-276. [ Links ]
32. Gómez-Moreno G, Cañete-Sánchez ME, Guardia J, Castillo-Naveros T, Calvo-Guirado JL. Orthodontic management in patients with haemophilia. About two clinical cases.Med Oral Patol Oral Cir Bucal. 2010;15(3):463-466. [ Links ]
33. Nematullah A, Alabousi A, Blanas N, Douketis JD, Sutherland SE. Dental surgery for patients on anticoagulant therapy with warfarin: a systematic review and meta-analysis. J Can Dent Assoc,2009;75(1):41-50. [ Links ]
34. El Batawi HY. Minimizing the risk of perioperative bleending in child with hemophilia A during rehabilitation under generl anesthesia: A case report. Int J Clin Pediatric Dent. 2013;6(3):217-222. [ Links ]
35. Pedemonte C, Montini C, Castellón L. Manejo de pacientes en tratamiento con anticoagulantes orales previo a cirugía oral. Revista Odontológica Mexicana, 2005;9(4):171-177. http://new.medigraphic.com/cgi-bin/resumen.cgi?IDARTICULO=9445 [ Links ]
36. Givol N, Hirschhorn A, Lubetsky A, Bashari D, Kenet G. Oral surgery-associated postoperative bleeding in haemophilia patients - a tertiary centre's two decade experience. Haemophilia.2015;21(2):234-240. [ Links ]
37. Dudeja GP. Dudeja KK. Lakhanoal M. Ali S. Endodontic managment of haemophilic patient- A clinical perspective. J Clin Diag Res. 2014;8(7):zd17-zd18.
[ Links ]
38. Franchini M, Rossetti G, Tagliaferri A, Pattacini C, Pozzoli D, Lorenz C, et al. Dental procedures in adult patients with hereditary bleeding disorders: 10 years experience in three Italian Hemophilia Centers. Haemophilia.2005;11(5):504-509. [ Links ]
39. Van Galen KPM. Engelen ET. Mauser-Bunschoten EP. Van Es RJJ. Schutgens REG. Antifibrinolytic therapy for preventing oral bleeding in patients with haemophilia or Von Willebrand disease undergoing minor oral surgery of dental extraction (Review). Cochrane Database of systematic reviews 2015, Issue 12 Art No. CD011385. [ Links ]
40. Piot B. Et al. Managment of dental extarctions in patients with bleeding disorders. Oral. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2002;93:247-50. [ Links ]
41. Frachon X. Et al. Managment options for dental extarction in hemophiliacs: A study of 55 extractions (2000-2002). Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2005;99:270-5. [ Links ]
42. Hsieh JT. Klein K. Batstone M. Ten-year study of postoperative complications following dental extraction in patients with inherited bleeding disorders. J Oral Maxillofac Surg. 2017. http//dx.doi.org/10.1016/j.ijom2017.04016 [ Links ]
43. Piot B, Sigaud-Fiks M, Huet P, Fressinaud E, Trossaërt M, Mercier J. Management of dental extractions in patients with bleeding disorders. Oral Surgery, Oral Medicine, Oral Pathology, Oral Radiology and Endodontics.2002;93(3):247-250. [ Links ]
44. Djulbegovic B, Goldsmith GJ. Guidelines for management of hemophilia A and B [letter; comment]. Blood.1995;85(2):598-599. [ Links ]
45. Plug I, Mauser-Bunschoten EP, Bröcker-Vriends AHJT, Amstel HKP van, Bom JG van der, Diemen-Homan JEM van, et al. Bleeding in carriers of hemophilia. Blood.2006;108(1):52-56.
[ Links ]
46. Bastounis E, Pikoulis E, Leppäniemi A, Alexiou D, Tsigris C, Tsetis A. General surgery in haemophiliac patients. Postgrad Med J.2000;76(898):494-495. [ Links ]
47. Cahill MR, Colvin BT. Haemophilia. Postgrad Med J.1997;73(858):201-206. [ Links ]
48. Rick ME, Walsh CE, Key NS. Congenital Bleeding Disorders. Hematology. 2003;2003(1):559-574. [ Links ]
49. Pipe SW. Hemophilia: New Protein Therapeutics. Hematology. 2010;2010(1):203-209. [ Links ]
50. Philipp C. The Aging Patient with Hemophilia: Complications, Comorbidities, and Management Issues. Hematology.2010;2010(1):191-196. [ Links ]

