











 Espanhol (pdf)
Espanhol (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por email Citado por SciELO
Citado por SciELO  Citado por Google
Citado por Google  Similares em
SciELO
Similares em
SciELO  Similares em Google
Similares em Google 
 Permalink
PermalinkIntroducción
La percepción del dolor es una respuesta adaptativa a la presencia de eventos dañinos y potencialmente mortales 1,2. Se han clasificado los trastornos de la reactividad al dolor en congénitos y adquiridos 3. Las neuropatías hereditarias (HSAN, del inglés Hereditary Sensory and Autonomic Neuropathy) se clasifican en 5 tipos. El Tipo II se asocia a disfunción autonómica con severa alteración en la función sensitiva 4. En 1883, Morvan describió 7 casos de un nuevo síndrome, que se caracterizaba por analgesia de las extremidades 5. Actualmente se sabe que HSAN tipo II es un desorden autosómico recesivo, en el cual existe una pérdida de la discriminación dolorosa, temperatura, sensación de tacto, así como de la respuesta afectivo-emocional. Los genes implicados son el cromosoma 12p13.33 y HSN2, y alternativamente un exón empalmado del gen WNK1 6.
La prevalencia mundial de HSAN tipo II es desconocida y no tiene preferencia de sexo 7. A modo de comparación, la prevalencia general de las neuropatías motoras y sensoriales hereditarias estrechamente relacionadas (como la enfermedad de Charcot-Marie-Tooth) es del orden de 30: 100.000, y las neuropatías sensoriales y autonómicas hereditarias (del tipo I al V) ocurren con una frecuencia notablemente más baja 8.
Por lo anterior, es esencial, llevar a cabo el diagnóstico y tratamiento a una edad temprana en estos pacientes, debido al riesgo de trastornos en el crecimiento y automutilaciones. De ahí la importancia de dar a conocer las complicaciones que pueden ocurrir con el fin de que la comunidad médica detecte a tiempo a este tipo de pacientes. Este reporte, muestra el seguimiento de un paciente femenino con Neuropatía Autonómica y Sensorial Hereditaria Tipo II durante diez años, con énfasis en el abordaje de las complicaciones dentales que se presentaron.
Descripción del caso
Paciente femenino de 13 meses de edad fue llevada a consulta al Centro de Especialidades Odontológicas del Instituto Materno infantil del Estado de México (CEO-IMIEM); originaria y residente de una población rural, Santa María del Monte, localizada en el municipio de Zinacantepec, Estado de México, México; el motivo de la consulta fue por caries en los dientes anteriores superiores. A la inspección oral clínica se observaron labios y lengua irregulares con pérdida de continuidad del tejido, así como tejido cicatrizal. A la inspección general, eran notables las escaras en el rostro y las lesiones en los dedos de manos y pies, las cuales sus padres reportaron como auto-infligidas.
Antecedentes hereditarios
Al interrogatorio de los antecedentes hereditarios, los padres refirieron que la paciente fue producto de la tercera gestación, de embarazo a término, con atención intrahospitalaria y trabajo de parto prolongado. Al nacer, la paciente no exhibió llanto ni respiración espontáneos, pesó 3300g, el tipo sanguíneo es Rh O+, se desconoce el APGAR neonatal, con tamiz metabólico sin alteraciones. Los eventos indicativos del desarrollo psicomotriz de la paciente todos se presentaron con retraso cronológico. Los padres no reportaron consanguineidad entre sí.
Evolución del caso
A los 3 días de edad presentó llanto inconsolable, por lo que los padres acudieron a un médico particular, el cual emitió el diagnóstico de síndrome no especificado y es referida a la Unidad Básica de Rehabilitación del Desarrollo Integral de la Familia (DIF) de Zinacantepec, donde recibió terapia durante los siguientes 3 años, observando mejoría, en cuanto a movilidad corporal. A los 3 meses de edad presentó cuadros de hipertermia sin causa aparente, alternados con hipotermia, sin manejo médico.
A los 2 años de edad, posterior a la aplicación de una vacuna no especificada presentó crisis convulsivas, por lo que fue hospitalizada 3 días, durante los cuales fue tratada con valproato de magnesio, y continuó con el fármaco de forma ambulatoria durante 3 años. A raíz de las convulsiones, no le fueron administradas las vacunas subsecuentes.
Al acudir por primera vez al servicio de odontología en el CEO-IMIEM, se detectó secreción purulenta en la zona del OD51 con luxación, por lo que se decidió realizar la exodoncia del mismo, en la radiografía se mostraba que además estaba involucrado el germen del OD11 (Fig. 1a, 1b). Nueve meses después, fue llevada a consulta de urgencia, por infección en la zona mandibular anterior, se extrajeron OD72 y el germen dentario OD41 (Fig. 1c y 1d). Posteriormente, al siguiente mes, se diagnosticó celulitis serosa en la zona maseterina y mandibular derecha (Fig.1f) por lo que se extrajo el OD85 (Fig.1h); adicionalmente se tomaron radiografías de la zona anterior (Fig.1e) y de la zona mandibular izquierda (Fig. 1g), así como panorámica (Fig. 1i). Se indicó farmacoterapia con amoxicilina y paracetamol (dosis estándar pediátrica).
A la edad de 2 años 1 mes, se extraen los OD74 y OD62, por presentar movilidad patológica, indicándose amoxicilina/ácido clavulánico (suspensión, 125/31,25mg/12horas por 5 días). Un mes después se extrae OD83 por la misma razón. Cuatro meses después acude a consulta de urgencia por movilidad en OD75, exhibe movilidad patológica y exudado purulento. Se realiza extracción y se continuó con el mismo esquema antibiótico. Se observaron los OD72 y OD82 con movilidad patológica, radiográficamente se observaba escaso soporte óseo, indicándose la exodoncia. Cuatro meses después acude con presencia de tejido granulomatoso en la zona mandibular derecha e izquierda; se indicó cefalexina (125mg/8 horas/5 días). Dos meses después acude a consulta de urgencia refiriendo inflamación de la zona inferior anterior, radiográficamente, (Fig.1g) no se observó ninguna alteración. Se limpia la zona y se prescribió dicloxacilina (suspensión, 125mg/6 horas/5 días). Un mes después acude presentando celulitis en la zona mandibular, se indicó clindamicina (suspensión, 150mg/8 horas/5 días), se solicitó interconsulta con servicio de Cirugía Maxilofacial. Ingresó a hospitalización con diagnóstico de osteomielitis, manejo de doble esquema antibiótico con una estancia de 35 días (Fig. 1h). Un mes después, presenta aumento de volumen a nivel del agujero mentoniano, en esta zona se retiró una espícula ósea (dimensiones: 20x6mm) y se indicó amoxicilina (suspensión). Un mes después presenta movilidad del OD36 por ausencia de soporte óseo y se realiza la exodoncia, prescribiéndose el mismo antibiótico.
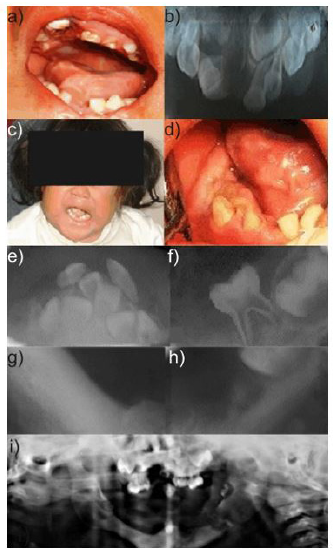
Figura 1 a) Inflamación intraoral de la zona anterosuperior b) Radiografía periapical órganos dentarios superiores, c) vista extraoral y d) vista intraoral de la zona mandibular derecha, se observa órgano dental en erupción, con tejido inflamatorio y secreción de material purulento. Radiografías periapicales de e) la zona anterior mandibular y f) zona mandibular derecha. g) y h) Radiografías periapicales mandibulares post-exodoncia y i) radiografía panorámica.
A la edad de 3 años 7 meses, acudió nuevamente a consulta de urgencia al CEO-IMIEM por presentar salida de exudado purulento. En el servicio de Cirugía Maxilofacial se extrajo germen del OD32 y se indicó dicloxacilina (suspensión, 125mg/6 horas/7 días). En la radiografía panorámica de seguimiento se encontró osteomielitis sin remisión, secuestro óseo del lado izquierdo con pérdida de solución de continuidad. (Fig.1h). A los 11 meses del diagnóstico de osteomielitis acudió a consulta y se extrajo el OD41 debido al escaso soporte óseo. Un mes después, continuaron los signos de osteomielitis, por lo que se realiza la mandibulectomía.
Al llegar a la edad preescolar, es evidente el retraso mental que padece y a consecuencia de esto, no acude a ninguna escuela. A los 5 años de edad, sufrió una fractura de tobillo izquierdo sin manejo médico, desconociéndose el mecanismo de lesión. La última convulsión registrada ocurrió a los 8 años de edad y fue llevada a Urgencias del Hospital del Niño (Toluca, Estado de México), donde se le administró ácido valproico (600mg/8horas).
En la Unidad del DIF, Zinacantepec, se le realizó una valoración neurológica, con diagnóstico presuntivo de epilepsia sintomática derivada de encefalopatía prenatal o probable HSAN, por lo cual es remitida al Centro de Rehabilitación y Educación Especial (CREE), Toluca, en donde se le realizó un electromiograma, el reporte del estudio fue neuroconducción motora y sensorial anormal, alteraciones que sugerían una neuropatía mixta crónica. En la exploración física, se registró que la paciente exhibía edad y talla menores a la edad cronológica. Por otra parte, ambos ojos presentaban opacidades cornales, sin reflejos pupilares, con seguimiento visual, distinguiendo sombras a distancia de 15 cm y presencia de úlceras corneales centrales vascularizadas (Fig, 2a), la cara presentaba tejido cicatrizal producto de la mandibuloctomia (Fig, 2b y 2c) y cráneo normocéfalo (Fig. 2d y 2e). Se observó en los miembros superiores, – de los reflejos bicipital, tricipital, estilorradial y automutilación de la parte distal de las falanges de los 10 dedos de ambas manos. Durante la estimulación al dolor los miembros superiores e inferiores, no se obtuvo respuesta, la sensibilidad superficial conservada, pero con sensibilidad profunda alterada. El pie izquierdo, se observó el antepie en supinación y pie equino; por lo que se indicaron tomas radiográficas de ambos pies (Fig. 2f, 2g y 2h). Otros órganos y sistemas no mostraban alteraciones. Posteriormente, la paciente fue referida al servicio de genética del Hospital del Niño donde los diagnósticos presuntivos fueron Síndrome de Moebius o HSAN, a descartar agenesia del cuerpo calloso. En ese mismo año, la paciente ya de 8 años de edad fue finalmente diagnosticada con Neuropatía Autonómica y Sensorial Hereditaria Tipo II.
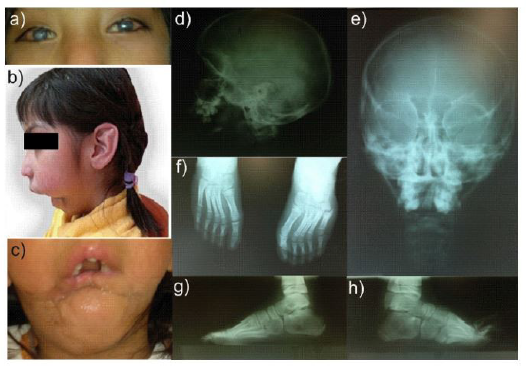
Figura 2 a) Opacidad corneal, b) vista frontal y c) lateral de la paciente. Radiografías d) lateral de cráneo y e) anteroposterior. Radiografías f) de ambos pies, g) pie izquierdo y h) pie derecho, se observa posición inadecuada de las estructuras óseas, debido a fracturas no tratadas.
A los 10 años de edad, reingresa al CEO-IMIEM por dolor en la zona posterior por erupción de órganos dentarios, así como dificultad alimentaria y respiratoria. Se tomaron radiografías panorámica y anteroposterior de cráneo (Fig. 3a y 3b) observando el deterioro y asimetría post-mandibulectomía. En la inspección clínica intraoral se observa colapso maxilar severo, gingivitis generalizada, dentición mixta, apiñamiento dental y lesiones cariosas (Fig. 3c) y, extraoralmente, es notorio el tejido cicatrizal en comisuras labiales y el labio inferior lo cual dificulta la apertura (Fig. 3d y 3e). Se realizaron la exodoncia de órganos dentales temporales y se rehabilitaron los permanentes remanentes. Se canalizó al servicio de Cirugía Maxilofacial en el Hospital para el Niño del IMIEM, en donde se realizó una traqueotomía y colocación de sonda gastropilórica para alimentación enteral. La paciente presentó buena evolución y volvió a tolerar la vía oral, egresando del servicio en buenas condiciones generales.
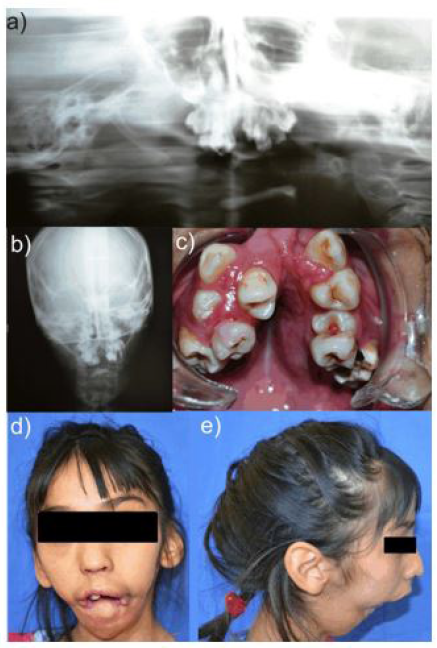
Discusión
Inicialmente en este caso, se planteó que podría tratarse del síndrome de Moebius, cuya incidencia es infrecuente (1:250000 nacidos vivos), caracterizada por parálisis facial congénita no progresiva unilateral o bilateral con alteraciones de la abducción ocular, anomalías orofaciales y defectos de las extremidades 9. En este caso, no había parálisis por lo que se descartó con un electromiograma. Otro diagnóstico presuntivo fue agenesia del cuerpo calloso, la frecuencia de esta anormalidad es de 1 en 4000 nacidos vivos, la relación masculina/femenino es 2:1, relacionándose con el síndrome de alcoholismo fetal. Clínicamente se observa un déficit neuropsiquiátrico severo, puede presentar alteración en áreas motoras de coordinación y tono muscular, falta de control de esfínteres, suelen presentar una alta tolerancia al dolor y en algunos casos, también se presentan crisis convulsivas 10,11. Esta anomalía se descartó con imagenología.
El signo más sobresaliente, en esta paciente, fue una progresión en la insensibilidad al dolor, evidente por sus lesiones. Entre los trastornos, que pueden causar alteraciones en la percepción del dolor, se encuentran las HSAN, las cuales se presentan a edad muy temprana. Particularmente, la HSAN-Tipo II, se caracteriza por una pérdida muy generalizada de sensaciones superficiales y profundas, pérdida de la sensación térmica y del tacto, incontinencia urinaria, disminución de la sudoración y reacción pupilar lenta a la luz 12,13,14. Dicha neuropatía tiene su expresión desde la edad pediátrica con lesiones auto-infligidas que pueden llegar a ser muy graves, incluso mutilantes. La ausencia de dolor conlleva la falta de mecanismos de protección ocasionando que microtraumatismos de repetición dañan la superficie articular y el hueso subcondral, produciendo una deformidad articular. Los pacientes presentan baja respuesta a tratamientos ortopédicos y frecuentes complicaciones post-quirúrgicas, así como pseudoartrosis, osteomielitis, artritis séptica, entre otras. Otra causa común de complicaciones, son las fracturas indoloras 12.
Para el diagnóstico existen tres pruebas relevantes: electrofisiológica, histopatológica y genética. La electrofisiología revela potenciales de acción nerviosos sensoriales reducidos o ausentes, velocidades de conducción nerviosas motoras preservadas o reducidas y potenciales de acción muscular compuestos reducidos. En el examen histopatológico del nervio sural se nota la ausencia pronunciada de fibras mielinizadas (pequeñas) y disminución de las no mielinizadas 3,8,15. Por último, las pruebas genéticas moleculares, que muestran la expresión de alguno de los cuatro tipos de HSAN Tipo II, y los genes asociados HSAN2A, HSAN2B, HSAN2C y HSAN2D 8.
Se desconoce un tratamiento etiológico, siendo el manejo sintomático la base del abordaje, con un equipo multidisciplinario, que brinde seguimiento de las complicaciones asociadas 16. Los individuos afectados en su mayoría tienen una inteligencia normal. Sin embargo, se han reportado casos en los cuales los pacientes padecen retraso mental 17. como en el presente caso, lo cual complica mucho la prevención de lesiones en estos pacientes. Un aspecto prevenible hubiera sido el colapso maxilar severo. que sufrió, sin embargo, la condición de la paciente no permitía el indicar un guarda oclusal. La falta de seguimiento en las citas no permitió llevar a cabo otro plan de tratamiento y/o prevención.
Dentro de las manifestaciones bucales se han descrito mutilación severa que inicia desde la erupción dentaria como mordedura de la lengua, los labios y la mucosa oral, luxación de órganos dentales, automutilación dentaria, desgaste dental 17. El tratamiento odontológico de elección consiste en eliminar las partes afiladas de los dientes y la colocación de guardas oclusales 13. El tratamiento debe ser conservador, anteriormente se consideraba necesaria la exodoncia de los dientes para prevenir daños a los tejidos bucales así como automutilación de extremidades, pero en la actualidad se pugna por tratamientos que preserven la calidad de vida 18.
Conclusiones
Cabe señalar que, la atención brindada a la paciente, en la mayoría de las ocasiones fue en los servicios de urgencia. A pesar de los esfuerzos por mantener a la paciente con la mejor calidad de vida posible, este caso muestra que la ausencia del diagnóstico temprano conduce a un pronóstico y condición de vida desfavorables para los individuos que sufren de esta anomalía genética.

