










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO  Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.19 no.3 Bogotá Sept. 2004
Poliposis adenomatosa familiar atenuada
Entidad controvertida
Jorge Mario Castro (1), Carlos E. Martínez (2), Jose A.Hormaza (3), Jaime Escobar C. (4), Lina Mateus (5).
(1) MD. Cirujano General, Universidad Nacional de Colombia, Fellow Cirugía y Endoscopia Colorectal. Hospital Militar Central. Universidad Militar Nueva Granada. Bogotá, D.C. Colombia.
(2) MD. Cirujano general y Colorrectal. Jefe del Servicio de Cirugía Colorrectal. Hospital Militar Central. Bogotá, D.C. Colombia.
(3) MD. Cirujano General y Colorrectal. Hospital Militar Central. Bogotá, D.C., Colombia.
(4) MD. Cirujano General y Gastrointestinal. Hospital Militar Central. Bogotá, D.C. Colombia.
(5) MD. Cirujano General. Fellow Cirugía y Endoscopia Colorrectal. Hospital Militar Central. Universidad Militar Nueva Granada. Servicio de Cirugía y Endoscopia Colorrectal, Hospital Militar Central. Bogotá, D.C., Colombia.
Resumen
La poliposis adenomatosa familiar es una patología recientemente descrita y hasta cierto punto desconocida por el grupo médico, se encuentran en la literatura principalmente reportes de casos y existe controversia sobre si es una patología propiamente dicha o la podemos ubicar en la mitad de un espectro que iría de pólipos adenomatosos aislados hasta la poliposis adenomatosa familiar clásica y el cáncer.
Los grupos que la consideran una entidad separada, afirman que es una variante de la poliposis adenomatosa familiar clásica, en la cual la expresión fenotípica es menor en lo referente al número de pólipos. Se ha identificado la mutación del gen APC responsable de su aparición y se ha tipificado el lugar específico del gen donde se produce, el número de pólipos es menor de 100, se ubican principalmente proximales al ángulo esplénico y el estudio histológico muestra con mayor frecuencia adenomas tubulares con displasia de bajo grado. El tratamiento puede ir desde la polipectomía hasta la colectomía total.
A propósito de algunos casos manejados en nuestro servicio, la mayoría de ellos con polipectomía y seguimiento estricto, decidimos hacer una revisión de la literatura, destacando los aspectos más relevantes de su etiología, diagnóstico y tratamiento.
Palabras claves : Poliposis atenuada, poliposis adenomatosa familiar, síndromes poliposicos, polipectomía.
Summary
Attenuated Familial Adenomatous Polyposis is an entity poorly understood for medical groups, is a variant of Familial Adenomatous Polyposis, characterized by fewer colorectal polyps, begin of polyps and cancer in later age than classic variant, and predilection for proximal ubication. Colonoscopy shows multiple flat adenomas, and histopathology is characterized by tubular adenomas with displasia in high percentage.
Diagnosis is difficult for abscence of specific finding or genetic proube, must be based in clinical and endoscopy findings, and treatment varies from polypectomy to total colectomy.
In this review we present the history, etiology, clinical presentation, diagnosis, and treatment of this pathology, and the experience with this pathology in our sevice.
Key words : Attenuated polyposis, familial adenomatous polyposis, polyposis syndromes, polypectomy.
Los pólipos adenomatosos de colon representan un reto importante para el clínico ya que además de la sintomatología que producen por su presencia, tienen un potencial de malignización importante; en la génesis del cáncer colo-rectal está bien establecida la secuencia adenoma-carcinoma, además dependiendo del tamaño y de las características histológicas del adenoma este riesgo se aumenta de forma importante.
La poliposis adenomatosa familiar clásica es un síndrome hereditario en el cual se encuentran más de 100 pólipos adenomatosos en colon y recto, aunque en algunos casos se pueden presentar más de 1000 pólipos, tiene un patrón de herencia mendeliano autosómico dominante, con cerca de 100% de penetrancia Los pacientes que presentan este síndrome desarrollarán cáncer colo-rectal después de la cuarta década de la vida casi en 100% de los casos de no recibir tratamiento quirúrgico (1).
Existe un subgrupo de pacientes en el cual se encuentra un menor número de pólipos, predominancia por el colon derecho y aparición más tardía de pólipos y cáncer colo-rectal, esta variante se ha conocido como poliposis adenomatosa familiar atenuada (PAFA).
La PAFA es una entidad poco frecuente y por lo mismo desconocida para el cuerpo médico, se encuentran pocos reportes en la literatura, y los casos descritos son series pequeñas. A continuación se revisarán los aspectos más relevantes de la historia, etiología, diagnóstico, cuadro clínico y tratamiento, y se comentará la experiencia con esta patología en nuestro medio.
Historia
En 1985 se describió en la literatura el término adenoma plano. Muto et al (2), describieron la presencia de lesiones planas elevadas de la mucosa con predilección por el colon derecho, caracterizadas por lesiones no mayores al doble del diámetro de la mucosa normal, con superficie rojiza y centro umbilicado, estas lesiones generalmente son menores de 1 cm, el estudio histológico muestra adenomas tubulares de diseminación lateral con alta incidencia de displasia.
Posteriormente, se encuentran algunos reportes de Henry Lynch, donde se describe una familia con cáncer hereditario no polipósico con adenomas planos ubicados en el lado derecho. Más adelante se reporta el patrón de transmisión autosómico dominante, y se anota también que la edad de aparición de los pólipos y del cáncer colo-rectal es mas tardía que en la variante clásica de la poliposis adenomatosa familiar.
En 1995, Lynch et al concluyen que la PAFA es una variante fenotipicamente distinta de la poliposis adenomatosa familiar clásica (3,4).
Etiología
En la poliposis adenomatosa familiar Clásica, se ha descrito con claridad el componente genético que la produce: las mutaciones del gen APC, ubicado en el brazo largo del cromosoma 5, han sido tipificadas como las responsables de esta patología.
El gen APC, es un gen supresor tumoral formado por 15 exones y 2843 codones, su función es codificar para la proteína del Gen APC. La proteína del gen APC tiene un tamaño de 312 Kda, y esta es la que tiene funciones directas en la tumorigénesis.
La proteína APC tiene funciones estructurales y de señalización, a nivel estructural tiene importancia uniéndose a proteínas del citoesqueleto, dando estabilidad a la célula, participa también en la división celular uniéndose a los husos cromáticos durante la mitosis, funcionando en este lugar como un mecanismo de control (5,6).
Como proteína de señalización se ha demostrado su participación en la unión a otras proteínas, impidiendo su interacción con otros péptidos, específicamente en la Poliposis Adenomatosa Familiar Clásica se ha descrito la importancia de la proteína APC, ya que en condiciones normales, esta se une a la B-catenina y previene su interacción con el factor célula B/factor linfoide, ya que funciona como un regulador negativo de la B-catenina, produciendo Down Regulation, disminuyendo así la concentración citoplasmática de B-catenina, cuando la Proteína APC está mutada, no ocurre la unión a la B-catenina, llevando a que se aumente la concentración citoplasmática de B-catenina, y esta se asocie con el complejo factor célula B/Factor linfoide, ocasionando una secuencia de transcripción diferente del DNA, que va a estimular la transcripción de secuencias específicas durante el ciclo celular, llevando a proliferación celular anormal, este ha sido descrito por algunos autores como uno de los primero pasos en la carcinogénesis (7).
Se han descrito 34 mutaciones distintas del gen APC en la PAFA, los exones 3, 4 y 6 son los más comúnmente afectados, la mayoría se encuentra ubicada en el extremo 5´ del gen.
Las mutaciones descritas tienden a ser cambios en pares de bases que llevan mutaciones tipo pérdida de sentido, las cuales resultan en detención prematura de la lectura por la aparición anormal de codones de finalización.
La ubicación de la mutación del gen es la responsable de la diversa expresión fenotípica de este síndrome, es decir dependiendo del lugar del gen donde ocurran las mutaciones, las manifestaciones clínicas, particularmente el número de pólipos va a ser diferente, cuando las mutaciones ocurren en los extremos del gen hay menor número de pólipos y se produce la poliposis atenuada, mientras que cuando la mutación se ubica en el centro del gen hay mayor número de pólipos y se produce la variante clásica de la poliposis adenomatosa familiar (8,9).
Dada la diversidad en la expresión fenotípica del síndrome, surgen nuevas hipótesis en relación a su etiología y la posible importancia de factores ambientales en la aparición de una nueva mutación germinal, sin embargo esto no se ha podido demostrar con claridad, y es uno de los retos de la genética y la biología molecular para los próximos años.
Presentación clínica
Los pacientes con Poliposis Adenomatosa Familiar Atenuada se presentan con menos de 100 pólipos, generalmente entre 3 y 50, en fases iniciales son adenomas planos proximales al ángulo esplénico del colon, hasta el momento se desconoce su asociación con el adenoma aserrado.
En promedio, los adenomas y el cáncer se encuentran a la edad entre 44 y 56 años respectivamente, este diagnóstico se produce 10 a 15 años después que en la variante clásica de la poliposis adenomatosa familiar. El desconocimiento de esta entidad por los clínicos origina un retraso en el diagnóstico y seguimiento de estos pacientes (8).
Los cánceres colo-rectales en estos pacientes están acompañados de adenomas sincrónicos ( Figura 1).
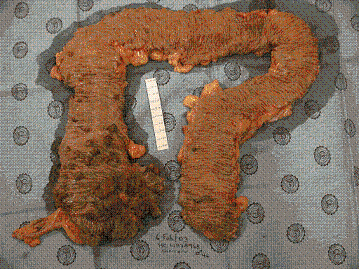
Figura 1. Pieza Quirúrgica de Colectomia Total en Poliposis Adenomatosa Familiar Atenuada, en la cual se observa mayor número de pólipos en el lado derecho.
Al igual que en la variante clásica, en la PAFA se encuentran manifestaciones extracolónicas de la enfermedad, estas incluyen adenomas gástricos o duodenales, tumores periampulares, y característicamente pólipos del fondo gástrico, estos últimos pueden preceder el desarrollo de adenomas colo-rectales. Ante el hallazgo incidental de pólipos en el fondo gástrico durante una endoscopia digestiva alta, está indicada la realización de la colonoscopia (10).
Otras lesiones asociadas son adenocarcinoma gástrico, cáncer de seno y hepatoblastoma.
Aunque otras lesiones como la hipertrofia congénita del epitelio pigmentario de la retina, osteomas y tumores desmoides son vistos con frecuencia en la variante clásica, aún no han sido descritos en la PAFA.
Diagnóstico
El diagnóstico de PAFA se basa en los hallazgos clínicos y endoscópicos del paciente, establecer con certeza el diagnóstico es más difícil que en la variante clásica, y esto se debe fundamentalmente a la variabilidad fenotípica de la PAFA, principalmente en lo referente al número de pólipos.
Los hallazgos colonoscópicos son los que nos orientan al diagnóstico, sumados a la edad, antecedentes personales y familiares del paciente. Es importante recordar que en ocasiones el paciente que nos consulta puede ser el portador de la mutación germinal inicial, y por lo tanto no tener antecedentes familiares. En la historia clínica siempre se debe buscar la presencia de sintomatología asociada a manifestaciones extracolónicas, y los estudios de extensión como endoscopia digestiva alta, y/o tránsito intestinal son parte fundamental de la aproximación diagnóstica.
Para la poliposis adenomatosa familiar clásica ya se han tipificado las mutaciones del gen APC más comunes, y existen en el mercado pruebas genéticas para detectar la mutación del gen; esta prueba es de mucha utilidad en personas de alto riesgo, o en pacientes con hallazgos endoscópicos especiales, ya que permiten detectar mutaciones antes que ocurra el cáncer, y tomar las medidas preventivas y de seguimiento.
La prueba más comúnmente utilizada es el test de truncacion de la proteína (PTT - Protein Test Truncation) esta prueba detecta 80% de las lesiones en la variante clásica, consiste en una electroforesis de proteínas en la cual se mide el tamaño de las proteínas, por lo tanto cuando hay mutación generalmente se encuentran las proteínas truncadas o acortadas y esto nos hace el diagnóstico, además siempre se recomienda el estudio genético para determinar la mutación germinal de cada familia (11).
En la PAFA no se dispone aún de una prueba genética con esta sensibilidad debido a que la ubicación de la mutación en los extremos del gen produce que el tamaño de las proteínas mutadas sea similar al de la proteína normal y la prueba inicial no detecta el acortamiento de la proteína. En individuos con alta sospecha de PAFA, lo ideal es realizar una secuenciación directa del DNA en los exones más frecuentemente comprometidos para detectar la mutación germinal. Es importante mencionar esto ya que una prueba (PTT) negativa en un paciente con PAFA no significa que el paciente no posea la mutación, lo que indica es que ésta se encuentra en otro lugar al detectado por la prueba.
Por todo lo anterior y hasta que no se disponga de pruebas genéticas sensibles y específicas para las mutaciones más frecuentes de la PAFA, es la colonoscopia la principal ayuda diagnóstica en Poliposis Atenuada. En ocasiones, ante pólipos pequeños puede ser difícil esclarecer un diagnóstico, en estos casos la experiencia del colonoscopista y la utilización de tinciones como indigo carmín son de gran ayuda ( Figura 2).
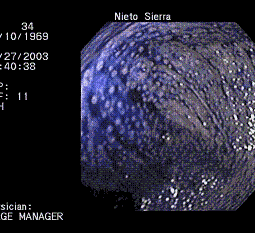
Figura 2. Imagen colonoscópica del ciego coloreado con indigo carmín en paciente con poliposis.
En el diagnóstico diferencial de la PAFA se debe mencionar el cáncer heredo-familiar no polipósico, (Síndrome de Lynch), ya que algunos casos de PAFA pueden presentar un número escaso de pólipos y hacer difícil el diagnóstico.
Similitudes entre síndrome de Lynch y poliposis atenuada
Las principales similitudes entre estas dos entidades se mencionan en la Tabla 1; principalmente ambas tienen predilección por el colon proximal, edad de aparición similar y antecedentes familiares, además en ambas entidades se pueden presentar tumores extracolónicos asociados lo cual hace dificil el diagnóstico, para estos casos se recomienda seguir los criterios de Ámsterdam y Bethesda para síndrome de Lynch, sin embargo en algunos pacientes por desconocimiento completo de la historia familiar, o sencillamente porque son portadores de la mutación germinal inicial estos criterios no hacen el diagnóstico con claridad y para estos casos se requiere una aproximación diagnóstica especial.
Considerando que la inestabilidad microsatelital se presenta en 85 - 100% de los casos de síndrome de Lynch, en casos de duda diagnóstica, esta puede ser la prueba inicial, en caso de resultar positiva se continúa el estudio para cáncer heredo-familiar no polipósico, en este caso se debe buscar la mutación de los genes MMR( Mismacth Repair Gene), y continuar el esquema recomendado (12-14).
Tabla 1. Similitudes entre poliposis atenuada y síndrome de Lynch.
| - Ubicación proximal de las lesiones - Edad de aparición de cáncer similar - Antecedentes familiares de cáncer colo-rectal - Tumores extracolónicos asociados |
Si la prueba de inestabilidad microsatelital es negativa, es poco probable que se trate de un síndrome de Lynch, y el caso se debe enfocar como una Poliposis Atenuada.
El PTT (Protein Test Truncation), es altamente sensible para Poliposis Adenomatosa Familiar Clásica, y poco sensible para Poliposis Atenuada.
Tratamiento
Aunque algunos autores proponen la realización de la prueba genética como inicio del tratamiento (7), en nuestro medio es la colonoscopia la que nos dará las pautas de manejo para cada caso (15).
En los casos en que se encuentren los pólipos de forma temprana, sin malignidad y el número de pólipos sea susceptible de manejo endoscópico se considera que la polipectomía endoscópica ( Figura 3), con seguimiento colonoscópico anual estricto es una buena opción terapéutica. Esto es apoyado por la observación clínica de pacientes con PAFA que han mostrado que la secuencia de pólipo a cáncer puede durar 10 años, este tiempo nos brinda una ventana grande sin cáncer para intentar el manejo no quirúrgico (16,17).
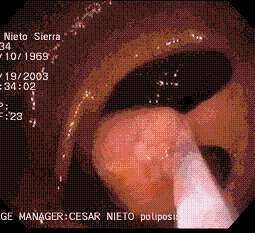
Figura 3. Polipectomía con asa en paciente con poliposis.
El número de pólipos para decidir manejo endoscópico es algo subjetivo, depende del lugar donde estemos y de la habilidad del endoscopista para la polipectomía; particularmente en nuestro servicio creemos que se puede intentar un manejo endoscópico del paciente en un gran número de casos, programando varias sesiones si es necesario, sin embargo algunos autores consideran que cuando hay gran compromiso del ciego la dificultad técnica hace preferible la cirugía.
Este manejo puede ser controvertido al tener que realizar una colonoscopia anual por el resto de la vida a los pacientes jóvenes de bajo riesgo quirúrgico-anestésico que adopten esta opción terapéutica, comparado con una cirugía definitiva, visto en términos económicos al cabo de 10 años de colonoscopias, sería menor el valor de la cirugía, sin embargo la morbi-mortalidad y la calidad de vida de los pacientes es mejor con la polipectomía.
En nuestro medio también se debe tener en cuenta el nivel cognitivo del paciente ya que este es un indicador indirecto de adherencia al tratamiento lo cual garantiza un adecuado seguimiento.
En casos en los cuales no sea técnicamente posible la polipectomia, o la posibilidad de seguimiento esté en duda, el paciente debe ser llevado a cirugía, en estos casos se debe realizar una colectomia total con bolsa ileal y anastomosis al recto, se recomienda la bolsa ileal sobre la anastomosis ileorectal convencional ya que la bolsa ileal permite que el paciente tenga de 1 a 3 deposiciones al día ( Figura 4) (18).
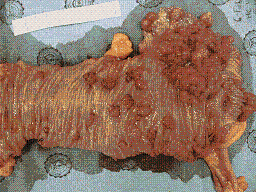
Figura 4. Adenocarcinoma moderadamente diferenciado de ciego, en paciente con Poliposis Atenuada.
Cuando el paciente se encuentra con cáncer, la cirugía es la misma, y se mantienen los preceptos de cirugía oncológica y valoración preoperatoria completa.
La proctocolectomia total no se indica en PAFA, debido a que por lo general, el compromiso del recto es poco frecuente, y en casos de pólipos a este nivel se puede ofrecer un seguimiento y manejo endoscópico con facilidad.
Estudios recientes muestran la utilización de anti-inflamatorios no esteroideos para el manejo de los pólipos evitando su progresión (19, 20), sin embargo, estos fármacos no modifican el potencial de malignización y la evidencia disponible al respecto no permite recomendar su utilización rutinaria con objetivo antitumoral (21,22)
Conclusión
La Poliposis Adenomatosa Familiar Atenuada, es una entidad nueva y hasta cierto punto desconocida, es una variante de la Poliposis Adenomatosa Familiar Clásica, en la cual hay menor número de pólipos, predilección por lesiones proximales, tiempo de aparición más tardío que en la variante clásica, su transmisión es autosómica dominante, y se han descrito hasta la actualidad 34 mutaciones diferentes del gen APC que la pueden producir. Es probable que en unos años no hablemos de poliposis clásica o Atenuada, sino de un espectro de lesiones y enfermedades del Gen APC, dependiendo de los avances de la genética y la biología molecular puede ser posible que más adelante una prueba genética nos indique la necesidad de una colectomía profiláctica para PAFA. Mientras tanto debemos tener en cuenta esta patología para determinar su incidencia y comportamiento en nuestro medio, con un diagnóstico y tratamiento precisos.
Referencias
1. Cao Y, Pieretti M, Marshall J, et al, Challenge in the Differentiation between Attenuated Familial adenomatous polyposis and Hereditary nonpolyposis Colorectal Cancer: Case report with Review of the literature. The American Journal of Gastroenterology; Vol 97,N 7:1822-1827 [ Links ]
2. Chrysos E, Athanasakis E, Vassilakis JS, Zoras O, Xynos E . Total Colectomyand J-Pouch Ileorectal anastomosis for obstructed tumours of the rectosigmoid junction. ANZ J Surg 2002 Feb; 72 (2):92-94. [ Links ]
3. Devita 1999, Prynciples of Oncology. [ Links ]
4. Giardiello FM, Brensinger JD, Petersen GM, et al. The use and interpretation of commercial APC gene testing for familial adenomatous polyposis. N Engl J Med 1997; 336:823-827. [ Links ]
5. Goldberg J, Perry B. Familial Adenomatous polyposis. Clinics in Colon and Rectal Surgery 2002; 15: 105-112. [ Links ]
6. Guillem JG, Smith AJ, Calle JP, Ruo L. Gastrointestinal polyposis syndromes. Curr Porbl Surg 1999;36:217-323. [ Links ]
7. Hernegger G, Moore H, Guillem J. Attenuated Familial Adenomatous Polyposis. Disease Colon Rectum 2002; 45:127-136. [ Links ]
8. Jagelman D. Familial Polyposis Coli, Current Therapy in Colon and Rectum Surgery; Fazio V. 1990. [ Links ]
9. Kelly K. Familial Adenomatous Polyposis; Current Surgical Therapy; Cameron 1998. [ Links ]
10. Leppert M, Burt R, Hughes JP. Genetic analysis of inherited predisposition to colon cancer in a family with a variable number of adenomatous polyps. N Eng J Med 1990;322:904-908. [ Links ]
11. Lynch HT, et al. Flat adenomas in a colon cancer-prone Kindred.J-Natl Cancer Inst. 1988;80:278-282. [ Links ]
12. Lynch HT, et al. Phenotypic variation in colorectal adenoma/cancer expression in two Families. Hereditary Flat adenoma. Cancer 1990; 66: 909-915. [ Links ]
13. Matsubara N, Isozaki H, Tanaka N. The Farhest 3 distal end APC mutation indentified in attenuated adenomatous polyposis coli extracolonic manifestations. Disease Colon Rectum 2000;43:720-721. [ Links ]
14. Muto T, Kamiya J, Sawada, et al. Small Flat adenoma of the large bowel with special reference to its clínico-pathological features. Disease Colon Rectum 1985;28:847-851. [ Links ]
15. Zwick A,Munir M, Ryan CK, et al. Gastric adenocarcinoma and dysplasia in Fundic Glands polyps of a patient with attenuated adenomatous polyposis coli. Gastroenterology 1997; 113: 659-663. [ Links ]
16. Rabelo R, Foulkes L, Gordon Ph, et al. Role of the Molecular Diagnostic testing in Familial Adenomatous polyposis and hereditary nonpolyposis colorectal cancer families. Disease Colon Rectum 2001;44.437-446. [ Links ]
17. Rubinfeld B, Souza B, Albert I, et al. Association of the APC gene product with B-catenin. Science 1993;262:1731-1734. [ Links ]
18. Niv Y, Fraser GM. Adenocarcinoma in the rectal segment in familial polyposis coli is not prevented by sulindac therapy. Gastroenterology 1994;107:854-857. [ Links ]
19. Nivatvongs S, Dorudi S. Colorectal polyps and their management. Colorectal Cancer; Churchill-Livingstone 1996. [ Links ]
20. Powell SM, Petersen GM, Krush AJ, et al. Molecular diagnosis of familial adenomatous polyposis. N Engl J Med 1993;329:1982-1987. [ Links ]
21. Steinbach G,Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med 2000;342:1946-1952. [ Links ]
22. Wallace MH, Frayling IM, Clark SK, Neale K, Phillips RK. Attenuated adenomatous polyposis coli: the role as ascertainment bias through failure to dye-spray at colonoscopy. Dis Colon Rectum 1999;42:1078-1080. [ Links ]

