










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.23 no.2 Bogotá Apr./June 2008
Fisiopatología de la pancreatitis aguda
Pathogenesis of acute pancreatitis
Jorge Iván Lizarazo Rodríguez (1)
(1) MD. Internista Gastroenterólogo Hospital Universitario La Samaritana. Docente Postgrado Universidad del Rosario. Bogotá, Colombia.
RESUMEN
La fisiopatología de la pancreatitis aguda, independientemente de las diferentes etiologías tiene como evento temprano la activación en el interior del acino pancreático de las enzimas pancreáticas que ocasionan el daño inicial de las células acinares pancreáticas. La magnitud del daño pancreático determina la severidad de la enfermedad y también la inducción de las respuestas inflamatorias y endoteliales a nivel local y sistémico, responsables de las complicaciones y el pronóstico de esta enfermedad.
Palabras clave
Fisiopatología, pancreatitis aguda, revisión.
SUMMARY
In the pathogenesis of acute pancreatitis, independent of the etiology, the activation of pancreatic enzymes inside the pancreatic acine is an early event. This causes initial injury to acinar pancreatic cells. The level of pancreatic injury determines the severity of the disease. It also determines whether or local and/or systemic endothelial inflammation reactions are triggered. These reactions can cause complications and are important for the prognosis of this medical condition.
Key words
Pathophysiology, Acute Pancreatitis, Review.
Fecha recibido: 20-05-08/ Fecha aceptado: 20-05-08
Con este artículo damos inicio al "Programa de Educación Médica Continuada", una iniciativa del autor, que ha sido puesta en marcha por el Comité Editorial y que cuenta con el respaldo de la Junta Directiva de la Asociación Colombiana de Gastroenterología. En cada número tendremos un tema seleccionado que será revisado en forma profunda y resumida con un enfoque didáctico. En el portal de la Asociación www.gastrocol.org el lector encontrará un cuestionario de evaluación, con preguntas relacionadas directamente con el texto publicado.
El páncreas es un órgano retroperitoneal con funciones exocrinas y endocrinas, la mayor parte de la glándula tiene funciones exocrinas, el 80% de las células son acinares y sólo el 2% son células de los islotes de Langerhans. Tiene un peso de 100 gramos y diariamente produce 500 cc de jugo pancreático compuesto por agua, electrolitos y enzimas digestivas. El páncreas tiene tres funciones fi siológicas generales:
a. Neutralizar el acido gástrico que ingresa al duodeno
b. Sintetizar y segregar enzimas digestivas
c. Liberar hormonas con funciones metabólicas.
En la presente revisión se profundizará la síntesis y secreción enzimática y su relación con el desarrollo de pancreatitis aguda (1-5).
SÍNTESIS Y SECRECIÓN DE LAS ENZIMAS PANCREÁTICAS
El acino pancreático está compuesto por 20 a 50 células acinares dispuestas en forma piramidal. En condiciones normales las células acinares sintetizan las enzimas pancreáticas en los polisomas que luego son transferidas al retículo endoplásmico rugoso y al aparato de Golgi, posteriormente son almacenados en forma de gránulos de zimógenos y secretados por exocitosis hacia el sistema ductal (figura 1), en tanto que las hidrolasas lisosomales después de sintetizadas son localizadas en forma separada de los zimógenos en los lisosomas (4).

Figura 1. Exocitosis en células acinares
La secreción pancreática de enzimas digestivas está controlada por sistemas neurales y hormonales, el duodeno es el principal órgano sensor para la secreción pancreática.
La secreción pancreática exocrina se produce continuamente tanto en la fase interdigestiva (ayuno) la cual es aproximadamente el 20% de la secreción máxima y en la fase digestiva con sus tres componentes básicos (cefálica, gástrica e intestinal); de éstas, en la fase intestinal la producción es máxima especialmente por la liberación de colecistoquinina (CCK) por las células duodenales en respuesta a ciertos aminoácidos (fenilalanina, metionina y valina) y ácidos grasos de la dieta. Una vez cesa este estímulo hay un control por retroalimentación de la secreción pancreática. El control de la secreción se produce por la inhibición de dos péptidos llamados péptido liberador de CCK (PLCCK) producido en las células intestinales y el péptido supervisor (PS) de origen pancreático que tienen la capacidad de estimular la liberación de CCK, estos péptidos posteriormente son degradados por las proteasas pancreáticas (tripsina, quimotripsina y elastasa) en la luz intestinal (1-5).
En períodos de ayuno las pequeñas cantidades de proteasas en la luz intestinal degradan la mayor parte de estos péptidos quedando solamente un estímulo muy bajo para la estimulación basal constante; posterior a la ingesta de alimentos las proteínas de la dieta requieren de las proteasas pancreáticas para su digestión y por lo tanto la concentración de proteasas libres para la degradación de estos pépticos disminuye, de manera que quedan sin degradarse y estimulan la liberación de CCK. Una vez digerido el alimento nuevamente las concentraciones de proteasas residuales que degradan estos pépticos aumentan y así se disminuye el estímulo para la secreción enzimática pancreática (5) (figura 2).

Figura 2. Activación de enzimas pancreáticas por enteroquinasa en ribete de cepillo.
Las enzimas pancreáticas que llegan al duodeno en forma activa son la lipasa y la amilasa, las demás enzimas se secretan en forma inactiva en forma de zimógeno, cuando éstas llegan al duodeno la enzima enteroquinasa presente en ribete en cepillo activa el tripsinógeno a tripsina. La tripsina activa los demás cimógenos a sus formas activas (1, 5) (figura 3).

Figura 3. Mecanismos de regulación de la secreción enzimática del páncreas.
FISIOPATOLOGÍA DE PANCREATITIS AGUDA
En condiciones normales las enzimas pancreáticas son activadas en la luz duodenal; existen varios mecanismos que protegen de la activación enzimática en el páncreas evitando su activación dentro del páncreas ocasionando pancreatitis; estos mecanismos son:
Las enzimas se almacenan en forma de gránulos de zimógeno
Las enzimas se secretan en forma inactiva
La enzima que activa los zimógenos se encuentra fuera del páncreas (Enteroquinasa duodenal)
Las células acinares producen inhibidores de tripsina como la serina proteasa inhibidor Kazal tipo 1 (SPINK1)
El gradiente de presión favorece el flujo de jugo pancreático hacia el duodeno
Las bajas concentraciones de calcio ionizado intracelular.
ACTIVACIÓN DE ENZIMAS PANCREÁTICAS
Estudios experimentales han encontrado que el bloqueo de la secreción enzimática y la activación del tripsinógeno y otros cimógenos son eventos que se encuentran en forma temprana hasta 10 minutos de iniciada la pancreatitis, lo cual sugiere fuertemente que este es el evento inicial y no resultado de ésta. Además de la activación de los zimógenos, se requiere que éstos permanezcan dentro de las células acinares después de ser activados para iniciar el daño celular.
Se han planteado varias teorías para la activación intraacinar de cimógenos:
Hipótesis de colocalización: plantea que el evento inicial es el bloqueo de la excreción de los zimógenos. Debido a que la síntesis es continua al bloquearse su excreción hay una acumulación progresiva de zimógenos que finalmente trae como consecuencia la aproximación y la fusión entre los gránulos de cimógeno y las enzimas lisosomales en un proceso denominado colocalización, esto conduce a que las enzimas lisosomales como la catepsina B activen los zimógenos a tripsina (3, 4). Esta enzima inicia el daño acinar pancreático. La retención de los gránulos de zimógeno posiblemente se debe a una alteración en la red de filamentos de actina que facilitan las funciones de secreción. Otra hipótesis plantea que la inhibición de la secreción se debe a una alteración de los procesos de secreción relacionada con proteínas denominadas SNARE localizadas en la membrana celular (3).
Otras hipótesis diferentes a la colocalización de los zimógenos incluyen la activación de éstos por los polimorfonucleares neutrófilos y otra también ha planteado que la elevación del calcio intracelular es el evento inicial en la activación de los zimógenos. En la actualidad existe evidencia de que el aumento del calcio intracelular es necesario pero no suficiente para activar el tripsinógeno e inducir pancreatitis.
FACTORES INTRACELULARES PROTECTORES
Existe una familia de proteínas que protege la célula contra mediadores inflamatorios y tóxicos, se denominan proteínas calientes de shock (heat shock proteins), estas proteínas son sintetizadas en condiciones de estrés celular, dos proteínas (HSP27 y HPS70) se han encontrado aumentadas en modelos de pancreatitis, se ha planteado que estas proteínas pueden atenuar el fenómeno de colocalización y evitar el aumento intracelular de calcio necesario para la activación de zimógenos aminorando la severidad de la pancreatitis (3).
Otro mecanismo protector es la alfa 1 antiproteasa que capta las proteasas y las transfiere a la alfa 2 macroglobulina que es la principal proteína inhibidora de proteasas circulante. En casos de liberación masiva de proteasas este sistema se satura y no logra inactivarlas (4).
RESPUESTAS INFLAMATORIAS EN PANCREATITIS AGUDA
La injuria inicial sobre las células acinares pancreáticas induce la síntesis y liberación de citoquinas que aumentan el reclutamiento de neutrófilos y macrófagos que a su vez aumentan la injuria pancreática y aumentan la producción de sustancias proinflamatorias como el factor de necrosis tumoral alfa y de interleuquinas (IL 1, IL2, IL6).
De estas citoquinas la IL 6 es una de las mejor estudiadas y caracterizada como inductor de reactante de fase aguda, estos mediadores también son responsables de la respuesta inflamatoria sistémica y complicaciones como el síndrome de dificultad respiratoria aguda del adulto (SDRA), relacionada con aumento de muerte temprana en pancreatitis, por el contrario la estimulación de la interleuquina 10 tiene efecto antiinflamatorio.
También se ha estudiado si la liberación de enzimas pancreáticas está relacionada con el compromiso inflamatorio sistémico, solamente al parecer la elastasa en estudios experimentales se ha relacionado como causa de injuria pulmonar, posiblemente porque aumenta la producción de citoquinas.
FACTORES NEUROGÉNICOS
En modelos experimentales se ha encontrado que el sistema neural intrapancreático es estimulado para liberar sustancia P y el péptido relacionado con el gen de la calcitonina (CGPR) que aumentan la permeabilidad vascular y la inflamación.
FACTORES VASCULARES
Se han encontrado alteraciones vasculares tanto en grandes vasos como en la microcirculación durante la pancreatitis aguda. Estudios angiográficos han demostrado una alta frecuencia de vasoespasmo en pancreatitis severas y la relación de zonas de necrosis con sitios de vasoespasmo. También se han encontrado alteraciones en la microcirculación relacionadas con la necrosis pancreática. Los factores asociados con el compromiso microcirculatorio son la sustancia p, la endotelina 1 y la sintetasa de óxido nítrico.
La activación endotelial adicionalmente facilita la migración de leucocitos y aumenta la liberación de sustancias inflamatorias.
FISIOPATOLOGÍA DE LA MUERTE CELULAR EN PANCREATITIS AGUDA
La necrosis pancreática es uno de los factores más importantes como predictor de pronóstico en pancreatitis aguda. Las investigaciones se dirigen a formas de prevenir o frenar la necrosis y así evitar el desarrollo de pancreatitis severa (figura 4).
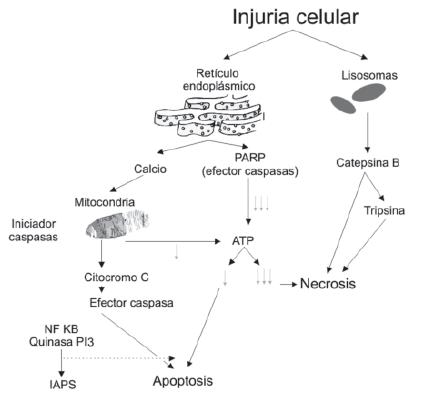
Figura 4. Mecanismos de muerte celular en pancreatitis aguda
En pancreatitis aguda se han identificado dos mecanismos de muerte celular que son la apoptosis y la necrosis: la apoptosis es denominada también como muerte celular programada, en condiciones fisiológicas controla la normal hemostasis de los tejidos, pero en condiciones patológicas como la pancreatitis aguda este tipo de muerte celular también se puede presentar. Las células que inician este proceso son reconocidas y eliminadas por los macrófagos, proceso en el que no hay rompimiento ni liberación del contenido celular al espacio extracelular por lo cual induce mínima inflamación.
La apoptosis se inicia con la activación de proteasas de cisteína denominadas caspasas, las cuales inician cambios a nivel mitocondrial cambiando la permeabilidad celular a través de la modificación del poro de permeabilidad de transición (PTP) para facilitar la liberación del citocromo C factor importante en esta vía. Adicionalmente en la apoptosis hay moderada depleción de ATP producido a nivel mitocondrial.
La necrosis es el segundo mecanismo de muerte celular en pancreatitis aguda, se presenta exclusivamente en condiciones patológicas, y produce disfunción mitocondrial severa, ruptura de la membrana plasmática y liberación de los constituyentes al espacio extracelular, se asocia con marcada respuesta inflamatoria; en la necrosis se encuentra una mayor disfunción mitocondrial manifestada por una mayor depleción de ATP, otros factores que regulan este tipo de muerte celular incluyen:
El PoliADP-ribosa polimerasa (PARP) que es activado por la ruptura del DNA durante la injuria celular y su activación utiliza ATP la cual aumenta su depleción.
El factor nuclear KB (NF-kB) y el sistema de fosfatidil inositol 3 quinasa (PI-3) que pueden promover la necrosis al aumentar la expresión del inhibidor de apoptosis (IAPs). En estudios experimentales la inhibición del NF-kB y el PI-3 podrían favorecer la vía de la apoptosis y no la de la necrosis, con mejoría de la severidad de la pancreatitis.
En condiciones fisiológicas el calcio se encuentra localizado en el retículo endoplástico y solamente se presentan pequeñas elevaciones transitorias de su concentración. En la pancreatitis hay una elevación de las concentraciones de calcio libre intracelular por su liberación desde el retículo endoplásmico aumentando en forma sostenida, esto ocasiona disfunción mitocondrial conduciendo a necrosis celular.
Finalmente la catepsina B, la mayor enzima lisosomal pancreática se ha demostrado que contribuye a la necrosis pancreática posiblemente a través de la conversión de tripsinógeno a tripsina.
En resumen, independientemente de la etiología de base, la severidad de la pancreatitis está relacionada a la magnitud del daño acinar pancreático inicial y la activación de la respuesta inflamatoria y endotelial que este daño genera y que ocasiona daños locales como la necrosis pancreática, la formación de pseudoquistes y abscesos y el compromiso sistémico y pulmonar por la liberación de mediadores inflamatorios en el páncreas o en otros órganos extrapancreáticos como el hígado.
REFERENCIAS
1. Lowe ME. The Structure and Function of Pancreatic Enzymes. Johnsons, Alpers, Chistenen, Jacobson, Walsh, eds. Physiology of the Gastrointestinal Tract. Vol 2. 3th Edition. Raven Press 1994. p. 1531-1542. [ Links ]
2. Frossard JL, Steer ML, Pastor CM. Acute pancreatitis. Lancet 2008; 371: 143-52. [ Links ]
3. Pandol SJ, Saluja AK, Imrie CW, Banks PA. Acute pancreatitis: Bench to the bedside. Gastroenterology 2007; 132: 1127-1151. [ Links ]
4. Sierra F, Torres D. Pancreatitis Aguda: una propuesta clínica basada en la "mejor" evidencia disponible (primera de 2 partes). Rev Colomb Gastroenterol 1999; 14: 170-180. [ Links ]
5. Forsmark CE. Enfermedades del Páncreas. En Wilcox CM, Ed. Digestive Diseases Self –Education Program [ Links ]

