










Services on Demand
Journal
Article
 Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.23 no.3 Bogotá July/Sept. 2008
Amiloidosis gastrointestinal (AG): Presentación de caso
Gastrointestinal amyloidosis
Roberto Rodríguez María, MD, (1), Hernán Eduardo Baquero Rodríguez, MD, (2), Eduardo Antonio Cruz Garrido, MD (2).
(1) Gastroenterología, UGASEND. Docente de Gastroenterología, postgrado de Medicina Interna Universidad Libre de Barranquilla. Barranquilla, Colombia.
(2) Residentes de Medicina Interna, Universidad Libre de Barranquilla. Barranquilla, Colombia.
Resumen
La amiloidosis es una enfermedad de etiología desconocida, que se caracteriza por el depósito de sustancia amorfa (amiloide), en los espacios extracelulares de diversos órganos y tejidos condicionando alteraciones funcionales y estructurales según la localización e intensidad del depósito. Alrededor del 75% de los pacientes que la padecen tienen una amiloidosis primaria, el 5% del total presenta amiloidosis secundaria, y menos del 5% desarrolla una amiloidosis familiar. Las manifestaciones clínicas son inespecíficas, determinadas por el órgano o el sistema afectado. El diagnóstico se basa en la sospecha clínica y la demostración de la sustancia amiloide en los tejidos. La evolución de la amiloidosis es difícil de comprobar debido a que casi nunca se conoce con precisión el inicio de la misma. En cuanto al tratamiento médico, se observa respuesta favorable con melfalán más prednisona; respecto al transplante de órganos, no se cuenta con un protocolo de aceptación universal, éste depende de cada caso, extensión y estadio evolutivo de la enfermedad.
A continuación presentamos un caso de amiolidosis primaria sistémica, en donde se encuentra comprometido el sistema gastrointestinal.
Palabras clave
Amiloidosis primaria sistémica, amiloidosis secundaria, insuficiencia renal, diarrea crónica, mieloma múltiple.
Abstract
The amyloidosis is a disease of unknown etiology, it is characterized for the deposit of amorphous substance (amyloide), in diverse organs and tissues extracell spaces determining functional an structural alterations according to the location and structural alterations according to the location and intensity of the deposit. About 75 % of the patients who suffer this disease have a primary amyloidosis, 5% of whole present suffer amyloidosis secondary, and less than the 5% develops a familiar amyloidosis. The clinical manifestations are not specifics, determinated for the organ or affected system. The diagnostic is based on the clinical suspicion and the proof of the amyloide substance on the tissues. It is really difficult to know the evolution of this disease because it is complicated to find the beginning of the same one. About the medical treatment they have observed favorable response with melfalan and prednisone. With regard to the organs transplant there is not protocol about universal acceptance it depends on every case, extension and evolutionary estate of the disease.
Now will present an amyloidosis primary systemic case, where one finds an affected gastrointestinal system.
Key words
Primary amyloidosis sistemic, secundary amyloidosis, renal failurie, Multiple myeloma, chronic diahrrea.
Fecha recibido: 11-03-08 Fecha aceptado: 12-08-08
Presentación de caso
Paciente femenino de 54 años de edad quien consulta inicialmente a medicina general por presentar cuadro de 4 meses de evolución, caracterizado por edema generalizado, astenia, adinamia, fatiga y pérdida de peso, asociándose además hace 1 mes a deposiciones líquidas 4-5 por día y dispepsia. Basado en esto y en la elevación ligera de creatinina el facultativo decide remitir al servicio de nefrología quien encuentra al examen físico:
- Regular estado general y músculo nutricional, palidez mucocutánea marcada.
- Signos vitales: TA: 140/90 mmHg; FC: 84 por minuto; FR: 19 por minuto; T: 37 grados centígrados; peso: 65 Kg
- CCC: edema facial, pupilas isocóricas normorreactivas a la luz; mucosa oral húmeda; no lesiones en cavidad oral o lengua; no macroglosia; no ingurgitación yugular.
- Tórax: simétrico, no tirajes.
- Cardíaco: ruidos cardíacos rítmicos, no soplos.
- Pulmones: claros, murmullo vesicular presente en ambos campos.
- Abdomen: peristalsis positivas, no circulación colateral, blando, onda ascítica positiva, hepatomegalia lisa, firme, no dolorosa a expensas de ambos lóbulos de 12-14 cms por debajo de reborde costal; no se palpa o percute esplenomegalia.
- Extremidades: edema grado II con fóvea en miembros superiores e inferiores; pulsos periféricos presentes.
- Sistema nervioso: consciente, orientado en sus tres esferas, fuerza muscular 5/5; reflejos osteotendinosos ++/++++.
- Piel: no se palpan adenopatías; no petequias o equimosis.
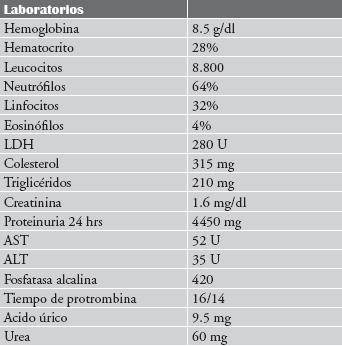
Basados en la clínica se ordena laboratorios y estudios de imágenes (tabla 1), dentro de los cuales se destacan anemia, elevación de fosfatasa alcalina y proteinuria en rango nefrótico. Además, se realizan ecografía abdominal confirmando la ascitis, hepatomegalia y encontrando aumento del tamaño de ambos riñones.
Se interconsulta al servicio de gastroenterología por persistencia de síntomas digestivos y le realizan esofagogastroduodenoscopia tomando biopsias duodenales para tinción de rojo congo (figuras 1, 2 y 3) y con estos hallazgos electroforesis de proteínas (figura 4) y biopsia de médula ósea reportando pico monoclonal e infiltración por plasmocitos atípicos, estableciéndose el diagnóstico definitivo de amiloidosis sistémica asociada a mieloma múltiple.
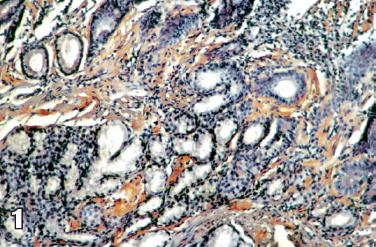
Figura 1 . Rojo Congo de tejido intestinal.
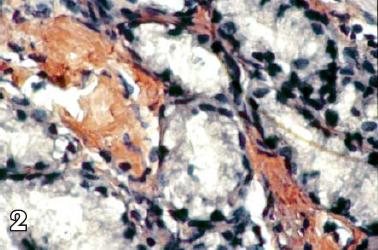
Figura 2 . Rojo Congo de tejido intestinal.
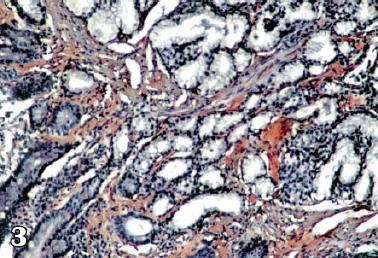
Figura 3 . Rojo Congo de tejido intestinal.
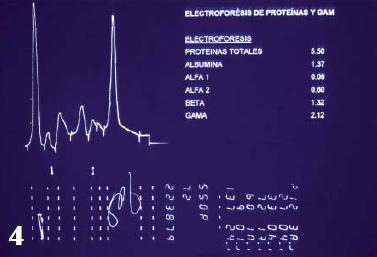
Figura 4 . Electrofóresis de proteinasRojo Congo de tejido intestinal.
Tabla 1. Estudios de laboratorio.
Discusión
La amiloidosis es una enfermedad sistémica o limitada a un órgano que resulta del depósito en los tejidos de la proteína amiloide (una proteína fibrilar resistente a la proteólisis con cadena B-plegada como estructura secundaria) (1).
En el siglo XIX los padres de la patología moderna Rudolf Virchow (1821-1902) y Carl F. Rokitansky (1808-1878), observaron en autopsias un proceso infiltrativo en los distintos órganos de la economía corporal el cual denominaron degeneración lardácea (del latín, laridum que significa semejante a la manteca); posteriormente al conservar dichos órganos en yodo, se comprobó que adquirían una coloración azul semejante al almidón, fue así como se comenzó a utilizar el término amilosis o amiloidosis (del griego Amylon, almidón. Eidos, forma) (2).
Existen varios subtipos de amiloidosis con diferentes unidades estructurales de proteína amiloidea. Estas incluyen amiloidosis primaria (también conocidas amiloidosis de cadenas ligeras), amiloidosis secundaria (también conocida como amiloidosis por amiloide A) y amiloidosis familiar. Las unidades estructurales de la proteína amiloidea para estos tres subtipos son fragmentos de cadenas ligeras de inmunoglobulina monoclonal, producto de interrupción de una proteína reactante de fase aguda conocida como amiloide A sérico y varias proteínas mutantes respectivamente (1).
La incidencia de amiloidosis primaria es difícil de definir precisamente. La incidencia en estados unidos ajustada a la edad es estimada entre 5,1 y 12,8 por millón de personas por año, siendo el 60% de los pacientes diagnosticados entre 50 y 70 años de edad y solamente el 10% menor de 50 años (3).
En la amiloidosis primaria la proteína fibrilar está constituida por cadenas ligeras de inmunoglobulina (más frecuentemente lambda que kappa en una relación 2:1). En más del 90% de casos se observa componente M en el suero. Es el tipo de amiloidosis que se observa en las discrasias de células plasmáticas (primaria o idiopática y la asociada a mieloma múltiple y macroglobulinemia) (4).
La amiloidosis primaria es caracterizada por depósitos de fibrillas amiloides en el corazón, produciendo falla cardíaca congestiva o arritmias; riñón, resultando en síndrome nefrótico o falla renal; el hígado, causando hepatomegalia y falla hepática colestásica progresiva; o el sistema neurológico, lo cual usualmente se manifiesta como neuropatía periférica sensoriomotora y disfunción autonómica. Muchos pacientes con amiloidosis primaria tienen depósitos demostrables histológicamente en el tracto gastrointestinal (5).
La amiloidosis primaria es el tipo más común de amiloidosis sistémica. La proteína amiloidogénica en amiloidosis primaria es una inmunoglobulina de cadena ligera o un fragmento de una cadena ligera que es producido por una población clonal de células plasmáticas en la medula ósea, ocurriendo en una asociación con el mieloma múltiple en el 10-15% de los pacientes (6).
Un hallazgo importante de todos los tipos de amiloidosis es el plegamiento anormal de una proteína que normalmente es soluble (7). En la amiloidosis primaria, el plegado anormal es el resultado de un evento proteolítico o una secuencia aminoacídica que da una cadena ligera termodinámicamente inestable y lleva a su autoagregación. Los agregados forman protofilamentos que se asocian dentro de la proteína fibrilar (8).
En todos los tipos de amiloidosis los glucosaminoglicanos, mitades de los proteoglicanos y el amiloide P sérico (SAP) (9) interactúan con las fibrillas amiloides, promoviendo la formación de fibrillas y la estabilidad en los tejidos (8). La disfunción de un órgano resulta de una disrupción de la arquitectura del tejido por los depósitos de amiloide. Sin embargo, incrementos en la evidencia indican que las proteínas precursoras amiloidogénicas o los precursores agregados tienen efectos citotóxicos directos que también contribuyen a las manifestaciones de la enfermedad (10).
Los genes de la región variable de las cadenas ligeras de la inmunoglobulina que son expresados menos frecuentemente en el repertorio normal, indican que las características de la codificación de la línea germinal pueden contribuir a la predisposición de ciertos subtipos de formas de amiloide de cadenas ligeras. La evidencia de antígenos selectivos en los genes de la región variable de las cadena ligeras de la inmunoglobulina como la homogeneidad de mutaciones somáticas soportan el concepto que la transformación monoclonal de varias células plasmáticas amiloidogénicas; ocurre después de la maduración de las células B y la selección clonal en el folículo linfoide (11). La asociación entre los genes usados en línea germinal de la región variable de las cadenas ligeras de la inmunoglobulina y la amiloidosis relacionada con el compromiso de un órgano ha sido descrita por varios grupos. Este tropismo por los órganos puede ser relacionado con las afinidades antigénicas de las cadenas ligeras (12).
Los órganos más frecuentemente afectados en la amiloidosis primaria son el riñón y el corazón (8); sin embargo, cualquier otro tejido como el cerebro puede ser comprometido. El riñón usualmente es comprometido presentándose como síndrome nefrótico con empeoramiento progresivo de la función renal (13). El depósito de amiloide en el corazón resulta en falla cardíaca rápidamente progresiva por cardiomiopatía restrictiva. Las paredes ventriculares son concéntricamente gruesas con un tamaño normal o reducido de la cavidad (14). El bajo voltaje en el electrocardiograma es encontrado en una alta proporción de pacientes y a menudo es asociada a un patrón de pseudoinfarto. La hepatomegalia es común y puede ocurrir como resultado de congestión por falla cardíaca derecha o infiltración de amiloide en el hígado. La hepatomegalia de la infiltración amiloide puede ser masiva y al examen físico el hígado suele ser como una roca dura y poco suave. La profunda elevación de la fosfatasa alcalina con ligera elevación de transaminasas es característica de la amiloidosis hepática porque la infiltración ocurre en los sinusoides (15). El compromiso del tracto gastrointestinal puede ser focal o difuso y los síntomas relacionados dependen del sitio y de la extensión. La macroglosia ocurre en el 10% de los pacientes y es virtualmente patognomónico; puede ser marcada causando obstrucción de la vía aérea, dificultad para tragar o apnea obstructiva del sueño (16). Otras características incluyen saciedad temprana, diarrea, náuseas crónicas, mala absorción y pérdida de peso (17). La amiloidosis gastrointestinal puede también presentarse con perforación intestinal o franco sangrado rectal (18). Ciertos síntomas como saciedad temprana y diarrea postprandrial explosiva a menudo reflejan disturbios de la motilidad gastrointestinal debido a neuropatía autonómica (19). La hepatomegalia está presente en aproximadamente un cuarto de los pacientes en el momento del diagnóstico y en la presencia de falla cardíaca puede no ser posible la diferenciación clínica entre infiltración amiloide y congestión venosa (20). La afectación renal y/o cardíaca constituye el principal evento clínico en la amiloidosis sistémica, determinando la afectación de estos órganos, frecuentemente, el pronóstico final del paciente (21). La polineuropatía por amiloidosis primaria puede incrementarse con un amplio rango de síntomas. Un poco más del 20% de los pacientes se presenta con síntomas de neuropatía periférica, más comúnmente parestesias, entumecimiento y debilidad muscular (22). La neuropatía sensorial es usualmente simétrica, usualmente afecta las extremidades inferiores y puede ser dolorosa; la neuropatía motora es rara (23).
La neuropatía autonómica es una característica mucho más seria, la cual puede darse hipotensión postural, impotencia sexual y disturbios de la motilidad intestinal que son usualmente asociados con algunos grados de neuropatía periférica (24).
La hemorragia es una importante manifestación de amiloidosis y puede ser un serio problema. Esto ocurre frecuentemente sobre un tercio de los pacientes y una coagulación anormal en alrededor de la mitad (24).
La manifestación más común del sangrado es la púrpura debido a la fragilidad vascular y como resultado del depósito de amiloide en el endotelio y puede ser una amenaza para la vida posterior a una biopsia hepática o renal. La púrpura periorbitaria (ojos de mapache) es particularmente característica (3).
La no especificidad y a menudo la naturaleza vaga de los síntomas que son asociados a la amiloidosis primaria frecuentemente lleva a una tardanza en el diagnóstico tal que la disfunción del órgano es muy avanzada en el momento en que se inicia el tratamiento. El diagnóstico de amiloidosis primaria debe considerarse en pacientes con proteinuria inexplicada, cardiomiopatía, neuropatía, hepatomegalia y en pacientes con mieloma múltiple que tienen manifestaciones atípicas.
El diagnóstico de amiloidosis primaria requiere demostración de una discrasia de células plasmáticas (25, 26). La demostración de los depósitos de amiloide en los tejidos mediante la birrefringencia verde manzana cuando es teñida con rojo congo y vista en microscopio polarizante. La aspiración con aguja fina de la grasa abdominal es un procedimiento simple que es positivo para los depósitos de amiloide en más del 70% de los pacientes con amiloidosis primaria (27, 28). Otros tejidos que permiten procedimientos de biopsia relativamente no invasivos son las glándulas salivales secundarias, encías, recto y piel. Sin embargo, la obtención de tejido de un órgano afectado en necesaria para el diagnóstico de amiloidosis.
Una vez que el diagnóstico en los tejidos se haya establecido, la confirmación de la enfermedad primaria requiere la demostración de una discrasia de células plasmáticas por biopsia de medula ósea de Lambda o Kappa, producidas por las células plasmáticas o por la presencia de unas cadenas ligeras monoclonales en el suero o la orina. La inmunofijación por electroforesis debe ser ordenada en el suero o la orina porque en contraste con el mieloma múltiple, la concentración de cadena ligeras monoclonales a menudo es también baja para ser detectada con electroforesis simple de proteínas.
Igual si una cadena ligera de inmunoglobulina es identificada en suero o la orina, la biopsia de medula ósea es mandatoria para evaluar la carga de células plasmáticas y excluir mieloma múltiple y otros desórdenes menos comunes que se pueden asociar a amiloidosis primaria tal como la macroglobulinemia de Waldenstrom (29, 30). Es importante reconocer que la presencia de una banda monoclonal en la inmunofijación sérica puede ser vista como un encuentro incidental en el 5-10% de los pacientes quienes están alrededor de los 70 años (Gammapatía monoclonal de significado incierto) (31).
El ensayo de cadenas ligeras libres en suero a menudo es normal en tales casos (32). Debido a la alta incidencia de Gammapatía monoclonal de significado incierto en individuos ancianos, pruebas adicionales deben ser hechas para excluir formas seniles o familiares de amiloidosis si el cuadro clínico es totalmente atípico para la enfermedad primaria. Tales pruebas incluyen inmunohistoquímica, inmunoflorescencia o microscopia electrónica de depósitos de amiloides para identificar la proteína amiloidogénica (33, 34) o pruebas genéticas para excluir formas familiares de amiloidosis (35).
La evaluación del grado de depósito amiloide es importante porque ayuda al pronóstico, plan terapéutico y determina la respuesta al tratamiento. Actualmente el examen físico y la biopsia de los tejidos son los métodos más usados para determinar el compromiso de un órgano. La escintigrafía usa sustancias que atan el amiloide siendo una herramienta útil en identificar órganos afectados. El pirofosfato de tecnecio 99 liga ávidamente a muchos tipos de amiloides y ha sido usado para identificar amiloide cardíaco (36). Sin embargo, la evaluación cuantitativa no es posible con esta prueba y es positiva solamente en pacientes con enfermedad severa, en quienes el ecocardiograma generalmente es diagnóstico. Resultados preliminares sugieren que el aprotinin sellado con tecnecio 99 puede ser más sensible para la imágenes de depósitos amiloides que el pirofosfato de tecnecio 99 (10). La cintigrafía cuantitativa también puede ser ordenada con el sello del componente SAP (serum amyloid protein) (10) una molécula que liga todos los tipos de amiloide (37). Las características ecocardiográficas de la amiloidosis cardíaca han sido bien descritas y razonablemente distintivas en enfermedad avanzada (38).
La paciente discutida corresponde a una amiloidosis primaria asociada a mieloma múltiple, la cual, como se describió anteriormente, es la forma más frecuente y debutó en la forma característica descrita en la literatura, por la edad de presentación, clínica por afección renal y gastrointestinal asociándose a elevación de fosfatasa alcalina, pico monoclonal, infiltración plasmocitaria de la médula ósea e incluso tinción de rojo congo, con lo cual se confirma diagnóstico.
Tratamiento
La terapéutica actual para la amiloidosis sistémica está basada en que la disfunción de un órgano mejora e incrementa la supervivencia si la síntesis del precursor de la proteína amiloidogénica es detenida. Por lo tanto, la principal terapia en la amiloidosis primaria es reducir rápidamente la fuente de cadenas ligeras monoclonales suprimiendo la discrasia de células plasmáticas subyacentes. La decisión sobre un régimen de tratamiento específico para pacientes individuales debe ser tomada en consideración en un equilibrio entre la eficacia del tratamiento anticipado y la tolerabilidad.
Sin importar el tratamiento específico dirigido contra la discrasia de células plasmáticas, el cuidado de soporte para disminuir los síntomas y la función de un órgano juega un papel importante en el manejo de esta enfermedad y requiere un cuidado especializado multidisciplinario.
Los síntomas gastrointestinales tales como saciedad temprana deben ser considerados en modalidad de selección. La diarrea es un problema común e incapacitante, en pacientes con compromiso del sistema nervioso autónomo. El octreotide disminuye la diarrea en muchos pacientes, pero la seudoobstrucción intestinal crónica usualmente es refractaria al tratamiento. La alimentación adecuada oral o intravenosa es mandatoria en pacientes desnutridos (39).
Los depósitos de amiloide permanecen en un estado de producción dinámica que varía marcadamente entre cada paciente, pero la regresión gradual de la amiloidosis primaria es a menudo vista cuando la producción de cadenas monoclonales es suprimida (40). El grado por el cual la enfermedad clonal necesita ser suprimida para producir beneficios clínicos varía sustancialmente entre pacientes y depende de muchos factores, notablemente de la producción de depósitos de amiloide. Los regímenes de quimioterapia usados en amiloidosis primaria están basados en aquellos que han probado ser efectivos en mieloma múltiple, aunque poco se sabe acerca de cualquier diferencia en la sensibilidad de las células plasmáticas clonales entre estos dos desórdenes. Los beneficios clínicos de la quimioterapia típicamente no ocurren por varios meses después de que las discrasias de células plasmáticas subyacentes han sido suprimidas (41). Los pacientes que reciben régimen de quimioterapia no viven mucho tiempo para mostrar el beneficio, por lo tanto es importante intentar suprimir la enfermedad clonal tan rápido como sea posible; sin embargo, regímenes más intensos de quimioterapia en pacientes con amiloidosis primaria están asociados con mucha mayor toxicidad relacionada con el tratamiento que han sido vistos en pacientes con mieloma. Esto es debido al daño de múltiples órganos lo cual puede no ser evidente clínicamente o por los resultados de rutina antes del tratamiento. La selección de un tratamiento apropiado individualizado para paciente es complicado y está compuesto además por la escasez de estudios aleatorizados para este desorden (3). La quimioterapia actual usada para la amiloidosis primaria puede ser clasificada como sigue:
- Dosis bajas: se utilizan como agente único melphalan o ciclofosfamida (con o sin prednisolona). Beneficios clínicos ocurren en solamente en 20 al 30% de los pacientes y después de una media de tratamiento de 12 meses. La combinación del régimen de quimioterapia usando VBMCP (vincristina, carmustina, mephalan, ciclofosfamida y prednisolona) no es más efectiva.
- Dosis intermedia: cursos mensuales de VAD (vincristina, adriamicina, dexametasona) y regímenes similares, o dosis intermedia de melphalan intravenoso (IDM) 25 mg por metro cuadrado con o sin dexametasona.
- Altas dosis: la terapia con altas dosis (HDT) con melphalan (100-200 mg por metro cuadrado) y transplante de células madre.
- Otras terapias: son pulsos de altas dosis de dexametasona y talidomida (con o sin dexametasona) con o sin otros agentes. Quimioterapia de dosis intermedia y de altas dosis parece ser clínicamente benéficas en más del 50% de los pacientes, pero no han sido comparadas en estudios aleatorizados uno con otro, ya sea con quimioterapia de bajas dosis o sin tratamiento (3).
- Terapias en investigación: un análogo de la talidomida, la lenalidomida y el bortezomib son activos en mieloma múltiple (42-45).
La habilidad de esta droga, en combinación con dexametasona reduce rápidamente la concentración de proteína monoclonal en mieloma múltiple haciéndolos una opción atractiva también para la amiliodosis primaria, aunque más datos en duración de la respuesta y toxicidad son necesarios. Sin embargo, estos estudios se encuentran sobre la vía.
Un derivado yodado de la doxorrubicina, el 4 yodo-4-deoxidoxorrubicina (IDOX) liga con alta afinidad las fibrillas amiloides y promueve su disgregación in vitro e in vivo en experimentos realizados en ratones en amiloidosis secundaria (46, 47). Las estrategias que combinan IDOX con quimioterapia para suprimir la producción del precursor y promover la resorción amiloide es una terapia racional que requiere investigación.
La terapia antifactor necrosis tumoral alfa, con el uso de etanercept, en 16 pacientes con amiloidosis primaria avanzada produce mejoría sintomática en la mayoría de los casos y la mitad tenía respuesta importante sobre todo en la macroglosia (48).
Pronóstico
El pronóstico es variable, pero es generalmente pobre si la amiloidosis primaria no es tratada. Los pacientes con amiloidosis primaria sistémica tienen una supervivencia mediana de uno a dos años (19). La historia natural varía con la extensión y órgano comprometido, pero poco menos del 5% de todos los pacientes con amiloidosis primaria sobre vive 10 o más años después del diagnóstico (49).
La evidencia clínica de compromiso cardíaco es un determinante importante de los resultados. La concentración sérica de la N-Terminal del péptido natiurético procerebral solo o en unión con los niveles cardíacos de troponina (50, 51) han mostrado ser un marcador sensible de disfunción cardíaca asociada a amiloidosis primaria y un fuerte predictor de supervivencia después del tratamiento agresivo. La alta circulación de niveles de cadenas ligeras libres han mostrado recientemente ser asociada con pobres resultados (52), la mayor reducción de los niveles después del tratamiento de la discrasias de células plasmáticas subyacentes son asociados con una reducción del N-Terminal del péptido natiurético procerebral y mejoría de la supervivencia (53).
Conclusiones
El diagnóstico temprano de la amiolidosis y la remisión apropiada tiene el potencial de mejorar los resultados para estos pacientes.
La amiloidosis primaria debe mantenerse como diagnóstico diferencial de pacientes evaluados por una variedad de síndromes, particularmente con proteinuria con rango nefrótico, cardiopatía no isquémica inexplicada, neuropatía periférica, hepatomegalia inexplicada o mieloma múltiple atípico, para mejorar la eficiencia en el diagnóstico. A pesar de estas mejoras en el tratamiento y diagnóstico de la amiloidosis primaria es básico continuar con los esfuerzos clínicos en la investigación de este campo para ayudar a mejorar los resultados con estos pacientes.
Referencias
1. Gertz Morie A. Diagnosing Primary Amyloidosis. Mayo Clinic. Proceedings 2002; 77: 1278-1279. [ Links ]
2. Lain Entralgo P. Historia de la medicina moderna y contemporánea. 2da edición. Ed. Científico Médica 1963; 2: 505-54. [ Links ]
3. United Kingdom Myeloma Forum. Guidelines on the diagnosis and menagement of Al amyloidosis. 2004; 125: 681-700. [ Links ]
4. R Sanmarti Sala, J Muñoz Gómez. Amiloidosis. Medicine 2001; 8(33): 1709-1714. [ Links ]
5. Robert A. Kyle, Morie A Gertz, Martha Q Lacy, Angela Dispenzieri. Localized AL amyloidosis of the colon an unrecognized entity. Amyloid: J. Protein folding disord 2003; 10: 36-41. [ Links ]
6. MF Khan, RH Falk. Amyloidosis. Postgrd Med J 2001; 77: 686-693. [ Links ]
7. Merlini G, Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349: 583-596. [ Links ]
8. Kisilevsky R. The relation of proteoglycans, serum amyloid P and apo E to amyloidosis current status. Amyloid 2000; 7: 23-25. [ Links ]
9. Aprile C, Marinone G, Saponaro R, Bonino C, Merlini G. Cardiac and pleuropulmonary AL amyloid imaging withtechnetium-99m labelled aprotinin. Eur J Nucl Med 1995; 22: 1393-1401. [ Links ]
10. Brenner DA, Jain M, Pimentel DR, Wang B, Connors LH,Skinner M, Apstein CS, Liao R: Human amyloidogenic light chains directly impair cardiomyocyte function through an increase in cellular oxidant stress. Circ Res 2004; 94: 1008-1010. [ Links ]
11. Abraham RS, Ballman KV, Dispenzieri A, Grill DE, Manske MK, Price-Troska TL, Paz NG, Gertz MA, Fonseca R. Functional gene expression analysis of clonal plasma cells identifies a unique molecular profile for light chain amyloidosis. Blood 2005; 105(2): 794-803. [ Links ]
12. Comenzo RL, Zhang Y, Martinez C, Osman K, Herrera GA: The tropism of organ involvement in primary systemicamyloidosis: Contributions of Ig V(L) germ line gene use and clonal plasma cell burden. Blood 2001; 98: 714-720. [ Links ]
13. Dember LM: Emerging treatment approaches for the systemic amyloidoses. Kidney Int 2005; 68: 1377-1390. [ Links ]
14. Kasper Dennis L, Braunwald Eugene, et al. Harrisons Principles of Internal Medicine. McGraw-Hill 2005; 310: 2024-2029. [ Links ]
15. Park MA, Mueller PS, Kyle RA, Larson DR, Plevak MF, Gertz MA. Primary (AL) hepatic amyloidosis: Clinical features and natural history in 98 patients. Medicine (Baltimore) 2003; 82: 291-298. [ Links ]
16. Falk RH, Comenzo RL, Skinner M. The systemic amyloidoses: current approaches to diagnosis and treatment. New England Journal of Medicine 1997; 337: 898-912. [ Links ]
17. Gertz MA. Amyloidosis: recognition, prognosis and conventional therapy. Hematology. American Society of Hematology Education Program Book. 1999, p. 333-347. [ Links ]
18. Kyle RA, Gertz MA. Primary systemic amyloidosis: clinical and laboratory features in 474 cases. Seminars in Hematology 1995; 32: 45-59. [ Links ]
19. Gillmore JD, Hawkins PN, Pepys MB. Amyloidosis: a review of recent diagnostic and therapeutic developments. British Journal of Haematology 1997; 99: 245-256. [ Links ]
20. Lovat LB, Persey MR, Madhoo S, Pepys MB, Hawkins PN. The liver in systemic amyloidosis: insights from 123I serum amyloid P component scintigraphy in 484 patients. Gut 1998; 42: 727-734. [ Links ]
21. Gavilán JC, Bermúdez FJ, Márquez A, Sánchez-Carrillo JJ, González-Santos P. Amiloidosis hepática como causa de colestasis severa intrahepática. An Med Interna (Madrid) 2003; 20: 25-27. [ Links ]
22. Rajkumar SV, Gertz MA, Kyle RA. Prognosis of patients with primary systemic amyloidosis who present with dominant neuropathy. American Journal of Medicine 1998; 104: 232-237. [ Links ]
23. Falk RH, Skinner M. The sytemic amyloidoses: an overview. Adv Intern Med 2000; 45: 107-37. [ Links ]
24. Mumford AD, ODonnell J, Gillmore JD, Manning RA, Hawkins PN, Laffan M. Bleeding symptoms and coagulation abnormalities in 337 patients with AL-amyloidosis. British Journal of Haematology 2000; 110: 454-460. [ Links ]
25. Westermark P, Benson MD, Buxbaum JN, Cohen AS, Frangione B, Ikeda S, Masters CL, Merlini G, Saraiva MJ, Sipe JD. Amyloid fibril protein nomenclature. Amyloid 2002; 9: 197-200. [ Links ]
26. Gertz MA, Dispenzieri A. Amyloidosis. Hematol Oncol Clin North Am 1999; 13: 1211-1220. [ Links ]
27. Libbey CA, Skinner M, Cohen AS. Use of abdominal fat tissue aspirate in the diagnosis of systemic amyloidosis. Arch Intern Med 1983; 143: 1549-1552. [ Links ]
28. Ansari-Lari MA, Ali SZ: Fine-needle aspiration of abdominal fat pad for amyloid detection: A clinically useful test? Diagn Cytopathol 2004; 30: 178-181. [ Links ]
29. Swan N, Skinner M, OHara CJ. Bone marrow core biopsy specimens in AL (primary) amyloidosis. A morphologic and immunohistochemical study of 100 cases. Am J Clin Pathol 2003; 120: 610-616. [ Links ]
30. Sanchorawala V, Blanchard E, Seldin DC, OHara C, Skinner M, Wright DG. AL amyloidosis associated with B-cell lymphoproliferative disorders: Frequency and treatment outcomes. Am J Hematol 2006; 81: 692-695. [ Links ]
31. Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Melton LJ 3rd: Long-term follow-up of 241 patients with monoclonal gammopathy of undetermined significance: The original Mayo Clinic series 25 years later. Mayo Clin Proc 2004; 79: 859-866. [ Links ]
32. Rajkumar SV, Kyle RA, Therneau TM, Clark RJ, Bradwell AR,n Melton LJ 3rd, Larson DR, Plevak MF, Katzmann JA: Presence of monoclonal free light chains in the serum predicts risk of progression in monoclonal gammopathy of undetermined significance. Br J Haematol 2004; 127: 308-310. [ Links ]
33. Arbustini E, Verga L, Concardi M, Palladini G, Obici L, Merlini G: Electron and immuno-electron microscopy of abdominal fat identifies and characterizes amyloid fibrils in suspected cardiac amyloidosis. Amyloid 2002; 9: 108-114. [ Links ]
34. OHara CJ, Falk RH. The diagnosis and typing of cardiac amyloidosis. Amyloid 2003; 10: 127-129. [ Links ]
35. Lachmann HJ, Booth DR, Booth SE, Bybee A, Gilbertson JA, Gillmore JD, Pepys MB, Hawkins PN: Misdiagnosis of hereditary amyloidosis as AL (primary) amyloidosis. N Engl J Med 2002; 346: 1786-1791. [ Links ]
36. Katzmann JA, Clark RJ, Abraham RS, Bryant S, Lymp JF, Bradwell AR, Kyle RA. Serum reference intervals and diagnostic ranges for free kappa and free lambda immunoglobulin light chains: Relative sensitivity for detection of monoclonal light chains. Clin Chem 2002; 48: 1437-1444. [ Links ]
37. Hawkins PN, Lavender JP, Pepys MB: Evaluation of systemic amyloidosis by scintigraphy with 123I-labeled serum amyloid P component. N Engl J Med 1990; 323: 508-513. [ Links ]
38. Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation 2005; 112: 2047-2060. [ Links ]
39. Vaishali Sanchorawala. Light-Chain (AL) Amyloidosis: Diagnosis and Treatment. Clin J Am Soc Nephrol 2006; 1: 1331-1341. [ Links ]
40. British Committee for Standards in Haematology. Clinical Haematology Task Force Guidelines on the provision of facilities for the care of adult patients with haematological malignancies (including leukaemia, lymphoma and severe bone marrow failure). Clinica and Laboratory Haematology 1995; 17: 3-10. [ Links ]
41. Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Thernau TM. A trial of three regimens for primary amyloidosis: colchicine, melphalan and prednisone and melphalan, prednisone and colchicine. New England Journal of Medicine 1997; 336: 1202-1207. [ Links ]
42. Richardson PG, Blood E, Mitsiades CS, Jagannath S, Zeldenrust SR, Alsina M, Schlossman RL, Rajkumar SV, et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 2006. [ Links ]
43. Rajkumar SV, Hayman SR, Lacy MQ, Dispenzieri A, Geyer SM, et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 2005; 106: 4050-4053. [ Links ]
44. Richardson PG, Barlogie B, Berenson J, Singhal S, Jagannath S, Irwin D, Rajkumar SV, et al. A phase 2 study of bortezomib in relapsed, refractory myeloma. N Engl J Med 2003; 348: 2609-2617. [ Links ]
45. Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadt-mauer EA, Facon T, et al. Bortezomib or high dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352: 2487-2498. [ Links ]
46. Palha JA, Ballinari D, Amboldi N, Cardoso I, Fernandes R,Bellotti V, Merlini G, Saraiva MJ. 4-Iodo-4-deoxydoxorubicindisrupts the fibrillar structure of transthyretin amyloid. Am J Pathol 2000; 156: 1919-1925. [ Links ]
47. Sebastiao MP, Merlini G, Saraiva MJ, Damas AM. The molecular interaction of 4-iodo-4-deoxydoxorubicin with Leu-55Pro transthyretin amyloid-like oligomer leading to disaggregation. Biochem J 2000; 1: 273-279. [ Links ]
48. Hussein MA, Juturi JV, Rybicki L, Lutton S, Murphy BR, Karam MA. Etanercept therapy in patients with advanced primary amyloidosis. Med Oncol 2003; 20: 283-290. [ Links ]
49. Kyle RA, Gertz MA, Greipp PR, Witzig TE, Lust JA, Lacy MQ, Thernau TM. Long-term survival (10 years or more) in 30 patients with AL amyloidosis. Blood 1999; 93, 1062-1066. [ Links ]
50. Palladini G, Campana C, Klersy C, Balduini A, Vadacca G,Perfetti V, Perlini S, Obici L, Ascari E, dEril GM, Moratti R, Merlini G: Serum N-terminal pro-brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003; 107: 2440-2445. [ Links ]
51. Dispenzieri A, Gertz MA, Kyle RA, Lacy MQ, Burritt MF, Therneau TM, McConnell JP, Litzow MR, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood 2004; 104: 1881-1887. [ Links ]
52. Dispenzieri A, Lacy MQ, Katzmann JA, Rajkumar SV, Abraham RS, et al. Absolute values of immunoglobulin free light chains are prognostic in patients with primary systemic amyloidosis undergoingperipheral blood stem cell transplant. Blood 2006; 107: 3378-3383. [ Links ]
53. Palladini G, Lavatelli F, Russo P, Perlini S, Perfetti V, Bosoni T, Obici L, Bradwell AR, DEril GM, Fogari R, Moratti R, Merlini G: Circulating amyloidogenic free light chains and serum N-terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL. Blood 2006; 107: 3854-3858. [ Links ]

