










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista colombiana de Gastroenterología
versión impresa ISSN 0120-9957versión On-line ISSN 2500-7440
Rev Col Gastroenterol v.23 n.4 Bogotá oct./dic. 2008
Patogénesis del cáncer de páncreas
Pathogenesis of pancreatic cancer
Jorge Iván Lizarazo Rodríguez (1)
(1) Médico internista gastroenterólogo Hospital Universitario de la Samaritana. Docente de Post grado Universidad del Rosario Bogotá Colombia.
Resumen
El adenocarcinoma pancreático es uno de los cánceres con mayor mortalidad, y la detección temprana y la sobrevida no han mejorado en la actualidad. Los estudios en las últimas dos décadas han demostrado que es una enfermedad fundamentalmente genética, causada por mutaciones hereditarias en línea germinal y/o mutaciones somáticas adquiridas en genes asociados con cáncer. Se han descrito lesiones precursoras de cáncer pancreático y alteraciones en varios genes importantes en el inicio y la progresión de esta enfermedad. Es posible que el mejor entendimiento de estas alteraciones permita en un futuro mejorar métodos de diagnóstico y de tratamiento.
Palabras clave
Cáncer de páncreas, genética, patogénesis, revisión.
Summary
Pancreatic Adenocarcinoma is a lethal disease for which early detection has not improved survival rates. Studies in the last two decades have shown that pancreatic cancer is fundamentally a genetic disease caused by inherited germ lines and acquired somatic mutations in genes associated with cancer. Multiple alterations of genes that are important in the progression of pancreatic cancer have been identified.
It is possible that better understanding of these alterations will facilitate identification of new biomarkers for early detection and may lead to novel drug targets.
Key words
Pancreatic cancer, genetics, pathogenesis, review.
Fecha recibido: 02-10-08 Fecha aceptado: 15-11-08
Con este artículo continuamos con el "Programa de Educación Médica Continuada", una iniciativa del autor, que ha sido puesta en marcha por el Comité Editorial y que cuenta con el respaldo de la Junta Directiva de la Asociación Colombiana de Gastroenterología. En el portal de la Asociación www.gastrocol.org el lector encontrará un cuestionario de evaluación, con preguntas relacionadas directamente con el texto publicado.
El cáncer pancreático es la cuarta causa de muerte por cáncer en Estados Unidos, en el mundo ocasiona aproximadamente 200.000 muertes, tiene una altísima mortalidad solamente del 0,4-4% sobreviven 5 años por lo cual la incidencia y la mortalidad son similares. En el momento del diagnóstico, aproximadamente el 80% de los casos son irresecables o tienen enfermedad metastásica. Los tratamientos con quimioterapia y radioterapia prácticamente no han modificado estas cifras.
El cáncer pancreático comprende un grupo de diferentes tumores; los principales son el adenocarcinoma ductal pancreático, los cistoadenocarcinomas, los tumores neuroendocrinos, el sarcoma pancreático, el carcinoma de células acinares y el linfoma. La presente revisión se basa sobre el adenocarcinoma ductal pancreático que corresponde a más del 80% de los casos (2). En la literatura se denomina como cáncer pancreático o adenocarcinoma pancreático. La lesión usualmente es una masa de color blanco amarillenta que compromete, en el 60% de los casos, la cabeza pancreática, en el 15% el cuerpo y cola y en el 20% en forma difusa la glándula; característicamente tiene una gran reacción desmoplásica, es un tumor muy agresivo con frecuente invasión linfática, vascular y a tejido perineural. En el estudio de las lesiones resecadas el 80% tiene compromiso ganglionar y en el momento del diagnóstico hasta el 80% de los pacientes tiene metástasis a distancia. Los sitios más frecuentes de metástasis son hígado 80%, peritoneo 60%, pulmón y pleura 50-70% y glándulas adrenales 25% (2).
Los estudios en las últimas dos décadas han mostrado que el cáncer pancreático es una enfermedad genética causada por mutaciones en la línea germinal y mutaciones somáticas adquiridas en genes asociados con cáncer (1). Existen una serie de anormalidades cromosómicas relacionadas con la fisiopatología y el desarrollo del cáncer pancreático, usualmente se presentan como una pérdida o ganancia de alelos en varios cromosomas en forma aleatorizada. Los cambios cromosómicos están relacionados con genes supresores de tumor, oncógenes o genes reparadores del DNA (1, 4).
Regulación del ciclo celular
Las células se dividen en respuesta a señales mediadas por factores de crecimiento externos y detienen su división en respuesta a factores de inhibidores que también actúan por múltiples señales.
Las sustancias inductoras externas pueden provenir de células vecinas (secreción paracrina) o de grupos celulares distantes (secreción endocrina). Estas sustancias actúan a nivel del punto de control G1, activan la síntesis de ciclinas y permiten el paso a la fase S.
Dentro de las sustancias inductoras del la proliferación celular encontramos los factores de crecimiento: FGF (Factor de crecimiento fibroblasto), PDGF (Factor de crecimiento plaquetario), EGF (Factor de crecimiento epidérmico).
En general, las células proliferan aumentado su contenido de moléculas de crecimiento y duplicando sus cromosomas para dividirse en dos células idénticas. La velocidad del ciclo celular es variable en los diferentes tejidos del organismo.
El ciclo de división celular tiene varias fases, la primera se denomina fase G1 en la que se repara cualquier daño en el DNA previamente al inicio de la fase de síntesis (fase S); en los mamíferos este punto de control es regulado por la proteína P53 que responde a la presencia de ADN dañado, esta fase tiene un punto restrictivo (Punto R) donde se define si se continúa el ciclo de división celular o se detiene. Este punto divide esta fase en temprana y tardía. La siguiente fase es la fase G2, donde se verifica que la replicación de ADN sea finalizada. La fase de síntesis es denominada fase S. Finalmente se completa la mitosis con la verificación de la correcta alineación de los cromosomas a través del huso mitótico y se asegura que cada célula hija reciba un juego de cromosomas (figura 1).
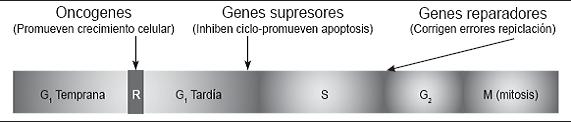
Figura 1. Función normal de genes relacionados con cáncer y ciclo celular
El ciclo de división celular en el carcinoma de páncreas como en otros tumores es un proceso complejo, es regulado por tres proteínas mayores que actúan para detener o permitir la progresión de la división celular. Estas proteínas son:
1. Las quinasas dependientes de ciclinas, en inglés (CDKs).
2. Las ciclinas.
3. Los inhibidores dependientes de ciclinas-quinasas (CKIs) (3).
Todas las células eucariotas tienen un reloj molecular que determina cuándo deben dividirse, programación dada por dos tipos de moléculas proteicas: Las ciclinas denominadas así porque alternan períodos de síntesis con períodos de degradación y las quinasas (CDK) que dependen de las ciclinas para su activación y tienen como función fosforilar moléculas cruciales para la división celular.
La unión de las ciclinas con las quinasas permite la fosforilación y la liberación del freno hacia la fase tardía G1 al desarmar un potente inhibidor de la progresión del ciclo formado por la proteína del retinoblastoma (pRB) denominada así porque fue en este tumor donde se descubrió y además, actúan sobre otros factores de transcripción inactivos (figura 2).
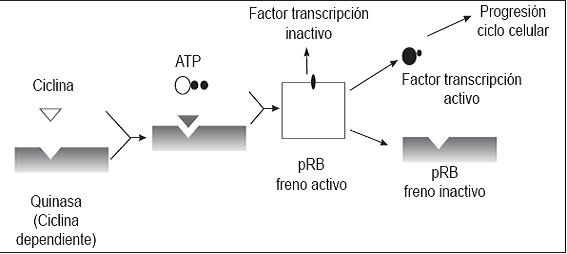
Figura 2. Regulación del ciclo celular
La activación de las ciclinas se presenta en forma secuencial (ciclina D-ciclina E-ciclina A-ciclina B (6).
Frenos de la división celular
Proteína P53 es sintetizada por la propia célula en respuesta a las alteraciones del ADN, se origina del gen p53 perteneciente a la categoría de genes supresores tumorales, en las células con alteraciones peligrosas para las células hijas la p53 se encarga de la muerte celular programada o apoptosis.
La proteína p53 también regula la expresión de otros genes de proteínas reguladoras como la p21 y la p16 (6).
La proteína reguladora de la proliferación celular más conocida es la denominada proteína Rb (derivada del gen Rb - gen supresor de tumor).
Los productos de oncógenes p21, p16, p27 actúan como inhibidores de quinasas (CKIs) al bloquear la hiperfosforilación del oncogen Rb por inactivación de CDK4-ciclina D y CDK2-ciclina E, así las células no pueden progresar de la fase G1 (figura 3) (3).

Figura 3. Interacciones moleculares del ciclo celular
Telómeros
Existe un mecanismo celular destinado a controlar el número de duplicaciones de una población celular, que se encuentra en los extremos de los cromosomas y se denomina telómero. Los telómeros, con cada división de los cromosomas, se van acortando y pierden aproximadamente entre 50-200 nucleótidos debido a la incapacidad de la DNA polimerasa para replicar la totalidad de los extremos del DNA, lo cual conduce a que las células entren en un proceso de envejecimiento y pierdan capacidad para dividirse.
En las células somáticas, con excepción de algunos tejidos de alta capacidad de autorregeneración no hay actividad de telomerasa. La inactividad de la telomerasa en las células normales ocasiona un acortamiento progresivo de los telómeros, causando inestabilidad cromosómica y aparición de diversos tipos de mutaciones, en esta situación es importante el reconocimiento que realiza la proteína p53 para inducir apoptosis de estas células.
Por el contrario, muchos tumores malignos expresan altos niveles de telomerasa, su reactivación al parecer es importante para el crecimiento tumoral (3).
Lesiones precursoras del cáncer pancreático
En el año 1999, se unificó la nomenclatura para clasificar las lesiones precursoras del cáncer pancreático denominadas como intraepitelial pancreática (PanINs). La clasificación considera cuatro grupos que reflejan los cambios histológicos progresivos desde el epitelio normal hasta terminar en carcinoma. El primer grado se divide en 1A lesión plana, y 1B lesión papilar, en este estadio hay ausencia de atipia o polaridad nuclear. El PanIN2, además de elementos papilares, tiene evidencia de atipias nucleares con infrecuentes mitosis y el PanIN3 tiene pérdida de la polaridad, atipias nucleares y mitosis frecuentes; esta lesión también se ha denominado carcinoma in situ (figura 4) (1, 2).
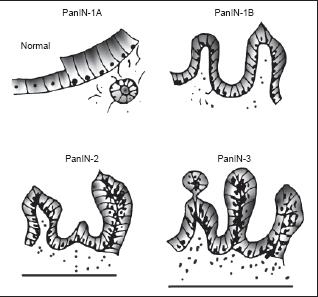
Figura 4. Neoplasia intraepitelial pancreática
esde el punto de vista genético está ampliamente aceptado que PanIN es la lesión precursora inicial del Ca de páncreas, cada estadio PanIN está asociado a modificaciones progresivas en oncógenes y genes supresores tumorales tales como: Ki-RAS (90-100%), p16 (90-95%), p53 50-85%, DPC4/SMAD4 (50%) y BRCA2 (10%). Se ha encontrado que la progresión del tumor requiere que esté alterado el Kras en combinación con el Ink4, debido a que el Ink4 y p53 frenan la progresión de PanIN a Ca de páncreas (7).
Bases genéticas
Todas las células tienen un DNA idéntico al DNA encontrado en el cigoto. Una mutación que se presente antes de este punto del desarrollo se denomina mutación en línea germinal, el defecto es transmitido a las demás generaciones y ocasiona el cáncer hereditario. Cuando la mutación se presenta en forma espontánea en el espermatozoide, óvulo o cigoto puede no encontrarse antecedente familiar previo pero si afectará la futura descendencia.
Hay mutaciones espontáneas "somáticas" que se presentan en células ya diferenciadas durante el desarrollo. Esta mutación origina una proliferación clonal de células. La mayoría de los cánceres esporádicos resultan por la acumulación de múltiples mutaciones somáticas en una célula (figura 5).
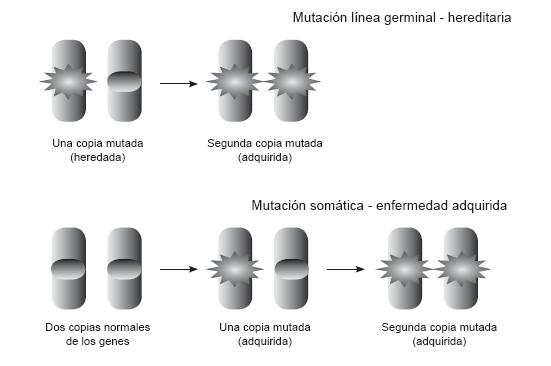
Figura 5. Pérdida de la función de genes supresores
Los genes comúnmente mutados en el cáncer humano pertenecen a una de estas tres clases oncógenes, genes supresores de tumor o genes reparadores.
Oncógenes
Son genes responsables de la estimulación y control de la proliferación celular, cuando mutan resultan en una proliferación descontrolada y el desarrollo de cáncer. Actúan de manera dominante, la mutación de una de las copias del gen es suficiente para su activación. Se pueden activar por diferentes mecanismos como mutaciones puntuales en el gen y la amplificación del mismo. En la actualidad existe un número creciente de oncógenes identificados en el cáncer de páncreas (1).
Genes supresores de tumorales
Fueron descritos por primera vez por Knudson en el retinoblastoma de niños. Los genes supresores tumorales se encargan de evitar el inicio del desarrollo tumoral, actúan en diferentes puntos del ciclo celular y al ser inactivados promueven el crecimiento tumoral. Son genes recesivos, es decir, las dos copias deben estar mutadas para que haya pérdida de la función. Existen diferentes mecanismos de inactivación de estos genes, por mutaciones intragénicas en un alelo y la pérdida del segundo alelo, o deleción de ambos alelos (deleción homocigota) y finalmente por hipermetilación del promotor del gen silenciador de la expresión genética. En cánceres esporádicos estas alteraciones pueden ser somáticas al adquirirse a lo largo de la vida o heredarse por vía germinal un alelo mutante y luego adquirir el segundo alelo mutado (figura 5) (1).
Genes reparadores DNA, en inglés DNA mismatch repair o MMR: regulan las enzimas que monitorizan el DNA formado y corrigen errores de replicación. Las mutaciones que afectan las dos copias del gen acumulan errores en el DNA, cuando afectan genes reguladores del crecimiento celular originan cáncer (figura 1).
Protoncógenes en cáncer pancreático
K-ras
K-ras (abreviación Kirsten Rat Sarcoma virus oncogen) es un protoncogeno localizado en el cromosoma 12p12.1, pertenece a la familia RAS que tiene tres protoncógenes H-ras, K-ras, N-ras. Están localizadas en la membrana plasmática. En cáncer pancreático las mutaciones K-ras se encuentran en el 74%-100%. Desde el punto de vista clínico esta mutación ha sido relacionada con disminución en la sobrevida (1).
Familia de genes Erb
A esta familia pertenecen el HER-2/neu, una familia de receptores de crecimiento epidérmico también conocido como ErbB2 y el factor de crecimiento epidérmico (EGF).
Los factores de crecimiento unidos a estos receptores promueven y activan el crecimiento y la diferenciación celular. La amplificación de estos protoncógenes lidera una proliferación incontrolada de las células. Se encuentran presentes entre el 16-65% de los Ca de páncreas, no se ha encontrado relación con el pronóstico y la sobrevida. En otros tumores como el cáncer de seno se relacionan con mal pronóstico pero también con una buena respuesta a anticuerpos monoclonales como el trantuzumab.
La sobreexpresión del EFG ha enfocado el interés en la utilización clínica de inhibidores como transtuzumab, erlotinib o cetuximab.
Notch 1 y Hedgehog
Se ha encontrado activación de algunas vías que normalmente participan en el desarrollo del cáncer pancreático. En los mamíferos existe una familia de proteínas de señal secretora denominada Hedgenog, que agrupa tres clases denominadas Sonic, Indian y Desert (Shh, Ihy, Dhh); tienen como función regular el crecimiento y el desarrollo durante la vida embrionaria de muchos órganos incluyendo el páncreas. La vía Hedgehog posteriormente se mantiene inhibida por una proteína supresora de tumor denominada Patched (PTC).
Otra vía es el Notch 1, que es un protoncógeno localizado en 9q34.3. Esta vía es importante en dirigir el destino y la proliferación celular durante el desarrollo embrionario. En etapas posteriores esta vía mantiene un balance entre la proliferación, la diferenciación y la apoptosis (1).
Estudios recientes han demostrado que la desregulación de una o más de estas vías y sus genes relacionados pueden iniciar cáncer en las células madre y la génesis tumoral. Tanto el Notch como el Hedgehog se han encontrado relacionados con el cáncer de páncreas por inducción de actividad de NF-KB que está presente en la mayoría de los tumores de páncreas. Algunos investigadores han encontrado activación de estas vías tanto en fases tempranas como avanzadas y metástasis de este tumor, adicionalmente la vía del Notch regula la neovascularización en el Ca de páncreas (4). Es probable que exista una comunicación entre las vías del Kras activado y las vías de señal Shh cooperando para preservar la apariencia de la lesiones PanIN; las fallas en esta asociación conducen a la formación de un tumor indiferenciado de características mesenquimales (7).
Genes supresores de tumores en cáncer de páncreas
P16/ink4
El gen p16 (también conocido como INK4, CDKN2MTS1) gen supresor de tumor tipo I se encuentra localizado en el cromosoma 9p21. Inhibe la interacción de la ciclina D con la CDK4 y la CDK6 frenando la división celular en la fase G1; las mutaciones en este gen promueven la proliferación celular y se encuentran en el 27-96% del Ca de páncreas. Desde el punto de vista clínico su relación con la sobrevida no ha sido consistente.
DPC 4 ó Smad4
Este gen supresor se encuentra en el cromosoma 18q. Es uno de los genes con más frecuencia mutados en el Ca de páncreas hasta en el 55%. La inactivación de este gen es relativamente específica del Ca de páncreas, es de baja incidencia en otros tumores pancreáticos y extrapancreáticos. Se han utilizado inmunomarcadores que muestran la pérdida de este gen por su especificidad como diagnóstico incluyendo el estudio de metástasis con sospecha de origen pancreático (1).
Factores de crecimiento
Se ha encontrado un aumento de la expresión de los factores de crecimiento en el Ca de páncreas, estos factores confieren ventajas en la proliferación de las células tumorales, favoreciendo un rápido crecimiento tumoral.
El factor de crecimiento epidérmico (EGFR), familia también es conocida como HER ó Erb B tiene 4 receptores. El tipo 2 se ha encontrado en lesiones precancerosas y tumores bien diferenciados.
Otro factor descrito en el Ca de páncreas es el factor de crecimiento endotelial (VEGF) que facilita el crecimiento vascular en la progresión tumoral.
El factor de crecimiento transformante beta (TGFB) generalmente es considerado como un supresor normal de la proliferación celular, sin embargo, sus vías de señal dan ventaja al crecimiento tumoral mediante sus efectos sobre el depósito de matriz extracelular, la promoción de marcadores mesenquimales, alteraciones del sistema inmune y la angiogénesis. El TGFB se ha encontrado sobreexpresado en Ca de páncreas al igual que su receptor. Adicionalmente se ha documentado una cooperación directa e indirecta con las vías de señal del Ras (7).
Acotamiento de los telómeros
Esta alteración puede ser la mayor causa de inestabilidad cromosomal en los tumores de páncreas, al parecer es un evento temprano y una de las principales causas de pérdida de la función de genes supresores de tumor y oncógenes. En la mayoría de los casos esta inestabilidad genómica es eliminada por la activación de la proteína p53, pero cuando hay mutaciones que afectan la p53 estos dos defectos cooperan en la génesis del cáncer en múltiples tejidos incluyendo el ca de páncreas (1).
Hiyama estudió la actividad de telomerasa en tumores pancreáticos y la comparó con entidades benignas como la pancreatitis crónica, encontró actividad de telomerasa en 95% de los carcinomas pancreáticos, en el 100% de lesiones con metástasis y en ninguna de las lesiones benignas. Se plantea la posible utilidad en el futuro como método diagnóstico (3).
Metaloproteinasas de matriz extracelular
Las MMPs pertenecen a una familia de por lo menos 20 miembros, actúan como enzimas dependientes de zinc. Las más conocidas son las colagenasas, estromelisinas y gelatinasas. Su principal rol es la degradación de los componentes de la matriz extracelular, participan en la remodelación y la cicatrización de los tejidos así como del desarrollo embrionario. Varias citoquinas, factores de crecimiento y factores de estrés mecánico pueden inducir la estimulación de la producción de MMPs.
Estas sustancias son de crucial importancia en la carcinogénesis porque además degradan la membrana basal y los componentes de la matriz extracelular favoreciendo los procesos de invasión tumoral y de metástasis.
Favorecen también la expresión de factores proangiogénicos tales como VEGF y bFGF, que contrarrestan los inhibidores angiogénicos tales como las trombospondinas, angiostatinas e INFs.
Debido a estas propiedades la inhibición de las MMPs es un blanco importante en el desarrollo de quimioterápicos contra el cáncer de páncreas.
Fibrogénesis
Los adenocarcinomas pancreáticos se caracterizan por presentar una incrementada fibrogénesis denominada "desmoplasia tumoral", se forma por un incremento en el tejido conectivo que infiltra y envuelve el tumor.
Las células estrelladas pancreáticas participan en la desmoplasia tumoral, se caracterizan por expresar alfa-SMA (actina de músculo liso) y por la síntesis de procolágeno alfa 1 T que son los principales componentes de la MEC (matriz extracelular) que constituyen la desmoplasia.
Las células estromales de estos tumores evidencian un aspecto morfológico dendrítico, expresan citofilamentos y una capacidad de síntesis de sustancias de matriz extracelular idénticos a los hallados en casos de pancreatitis crónica inducida por alcohol, lo cual sugiere unos mecanismos similares subyacentes al desarrollo de la fibrosis en la pancreatitis crónica y de la desmoplasia en el adenocarcinoma pancreático.
Existe además evidencia creciente de la relación que existe entre las células estrelladas pancreáticas y las células tumorales; por mecanismos parcialmente entendidos las células estrelladas pueden estimular el crecimiento tumoral, además pueden promover la invasión tumoral y posiblemente la angiogénesis (5).
Cáncer pancreático hereditario
Se han descrito algunos síndromes hereditarios que aumentan el riesgo de cáncer de páncreas, este grupo corresponde al 2-10% de los casos. Los individuos con uno, dos o tres familiares en primer grado con cáncer de páncreas tienen un aumento del riesgo de 6, 18 y 57 veces con relación a la población general. Se han descrito cinco síndromes hereditarios que se asocian con aumento del riesgo de cáncer pancreático:
1. Síndrome de cáncer familiar seno/ovario: se caracteriza por un aumento del riesgo de Ca de seno tanto en hombres como en mujeres, la mutación del gen BRCA2 que está en el cromosoma 13q, se encuentra en el 4-17% de Ca pancreático hereditario (1).
2. Síndrome FAMM: síndrome autosómico dominante caracterizado por múltiples nevus melanocíticos, aumento de riesgo de melanoma y carcinoma pancreático. La mutación en línea germinal del gen p16/CDKN2A está en el cromosoma 9p. Las personas afectadas tienen un riesgo estimado de 17% de Ca pancreático a los 75 años (1).
3. Síndrome de Peutz Jeghers: es una entidad autosómica dominante caracterizada por el desarrollo de pólipos hamartomatosos en tracto gastrointestinal, pigmentación mucocutánea y alto riesgo de cáncer. En el 50% de los casos la mutación en línea germinal compromete el gen STK11/LKB1, y tiene un riesgo de desarrollo de cáncer pancreático de 36% (1).
4. Síndrome de carcinoma colónico no polipoideo (HNPCC): es una entidad autosómica dominante que se asocia con múltiples formas de cáncer, el más frecuente es colorrectal, pero puede también asociarse con cáncer gástrico, endometrial, intestino delgado y pancreático, la mutaciones asociadas con Ca de páncreas son especialmente los genes hMLH1 y hMSH2, que son genes reparadores de DNA. Cuando uno de estos genes es inactivado, los cambios que ocurren conducen a inestabilidad microsatelital (MDI), aproximadamente el 4% de Ca de páncreas la tiene y característicamente son de tipo medular y tienen un mejor pronóstico (1, 2).
5. Pancreatitis hereditaria: se caracteriza por pancreatitis crónica con inicio en edades tempranas, la mutación puede ser dominante en línea germinal de genes como el PRSS1 o mutaciones recesivas en el gen SPINK 1; las personas afectadas tienen un riesgo de 40% de desarrollar cáncer pancreático a la edad de 70 años (1).
Estos síndromes solamente corresponden a menos del 20% de los casos familiares, y al 80% de los casos de cáncer pancreático familiar; la alteración genética de base permanece desconocida. Estos grupos de entidades son los que hasta el momento más se benefician de estudios de tamizaje para detección temprana, es posible que el estudio de estas familias mejore el conocimiento de la genética del cáncer pancreático (2).
Referencias
1. Kroorstra JBM, Hustinx SR, Offerhaus GJ. Pancreatic Carcinogenesis. Pancreatology 2008; 8: 110-125. [ Links ]
2. Winter JM, Maitra A, Yeo C. Genetics and pathology of pancreatic cancer. HPB (Oxford) 2006; 8(5): 324-336. [ Links ]
3. Saif MW, Karapanaglotou L, Syrigos K. Genetic alterations in pancreatic cancer. World J Gastroenterol 2007; 13(33): 4423-4430. [ Links ]
4. Strimpakos A, Saif MW. Pancreatic cancer: from molecular pathogenesis to targeted therapy. Cancer Metastasis Rev 2008; 27: 495-522. [ Links ]
5. Omary MB, Lugea A, Lowe AW, Pandol SJ. The pancreatic stellate cell: a star on the rise in pancreatic diseases. J Clin Invest 2007; 117: 50-59. [ Links ]
6. Hipertextos de Biología Molecular. Regulación del Ciclo Celular. http://fai.unne.edu.ar/biologia/cel_euca/regulacion.htm. [ Links ]
7. Rugstgi AK. The molecular pathogenesis of pancreatic cancer: clarifying a complex circuitry. Genes & Dev 2006; 20: 3049-53. [ Links ]

