










Servicios Personalizados
Revista
Articulo
 Español (pdf)
Español (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista colombiana de Gastroenterología
versión impresa ISSN 0120-9957versión On-line ISSN 2500-7440
Rev Col Gastroenterol v.24 n.2 Bogotá abr./jun. 2009
Síndrome de Peutz-Jeghers. Presentación de casos y revisión de la literatura
The Peutz-Jegherss Syndrome
John Ospina Nieto (1), Álvaro Pío Quintero (2)
(1) MD, MSCC, MSCG, MSCED, MSCH, MSCP, MFELAC. Cirujano Gastrointestinal y Endoscopista digestivo, Coordinador Gastroendoscopia Hospital Cardiovascular el Niño de Cundinamarca (Soacha) - Dispensario central del Ejercito. Bogotá, Colombia.
(2) MD, MSCC. Cirujano Gastrointestinal y Endoscopista digestivo. Hospital Universitario de la Samaritana. Dispensario Central del Ejercito. Bogotá, Colombia.
Fecha recibido: 24-07-08 Fecha aceptado: 26-03-09
RESUMEN
El síndrome de Peutz-Jeghers es una poliposis de baja incidencia que se caracteriza por la combinación de lesiones pigmentadas mucocutáneas y pólipos hamartomatosos del tracto gastrointestinal. Su patrón hereditario es autosómico dominante de penetración variable y en un alto porcentaje se asocia a mutaciones del gen STK11/LKB1, localizado en la banda 19p13.3. La presentación clínica varía desde paciente completamente asintomático hasta obstrucción intestinal y, a largo plazo, alto riesgo de cáncer gastrointestinal y extragastrointestinal y de síndrome de intestino corto (por resecciones múltiples). Este síndrome es de manejo sintomático en los eventos agudos y de seguimiento periódico con polipectomías endoscópicas de los pólipos más grandes con el fin de evitar la transformación hamartomatoma-adenoma-carcinoma y de detectar las lesiones tumorales tempranas. En el presente artículo presentamos dos pacientes, de 21 y 22 años, que ingresaron al Hospital Universitario de la Samaritana con cuadro de obstrucción intestinal y signos de abdomen agudo requiriendo resección intestinal y polipectomía quirúrgica con reporte de patología que mostró pólipos hamartomatosos. El seguimiento se realizó en el Hospital Cardiovascular del niño de Cundinamarca, con endoscopias, enteroscopias y polipectomías seriadas. Se presenta además una revisión acerca de esta infrecuente entidad y las recomendaciones vigentes de seguimiento a estos pacientes.
Palabras clave
Peutz-Jeghers, enteroscopia, polipectomía.
SUMMARY
The Peutz-Jegherss syndrome, it is a poliposis of low incident that characterizes for the combination of pigmented mucocutaneous and hamartoma polyps in the gastrointestinal tract. This is a dominant autosomic syndrome of changeable penetration and in a high percentage it is associates to mutations of the gene STK11/LKB1, located in the band 19p13.3. The clinical presentation changes from completely asymptomatic patient to, intestinal obstruction, high risk of gastrointestinal cancer and extra gastrointestinal neoplasm and of short intestine syndrome (for multiple resections). This syndrome is of symptomatic managing in the acute events and of periodic follow-up with polipectomy of the biggest polyps in order to avoid the transformation to neoplasm and of detecting the tumour early injuries. In the present article lets sense beforehand two patients who entered to the Hospital Universitario de la Samaritana with intestinal obstruction and signs of acute abdomen needing intestinal resection and polipectomy surgical with report of pathology that showed hamartom polyps. The follow-up I realize in the Hospital Cardiovascular del niño de Cundinamarca with endoscopy, enteroscopy and periodic polipectomy. One presents in addition a review over this infrequent entity and the in force recommendations of follow-up.
Key Words
Peutz-Jeghers, Enteroscopy, polipectomy.
El síndrome de Peutz-Jeghers, es una poliposis de baja incidencia que se caracteriza por la combinación de lesiones pigmentadas mucocutáneas y pólipos hamartomatosos del tracto gastrointestinal. Se considera una entidad bastante rara en nuestro medio, razón por la cual hemos decidido presentar una revisión del tema y unas guías de seguimiento para estos pacientes que tienen un riesgo de presentar neoplasias gastrointestinales y extragastrointestinales muy superiores al de la población general.
PRESENTACION DE CASOS
Caso 1
Paciente de 21 años con antecedente quirúrgico de laparotomía exploratoria por obstrucción intestinal por pólipos a la edad de 3 años y sin antecedentes familiares de importancia, quien ingresa en enero de 2006 al HUS con cuadro clínico de 4 horas de evolución de obstrucción intestinal; en la valoración inicial, abdomen con signos de irritación peritoneal, frecuencia cardíaca 100 x min, frecuencia respiratoria 20 x min, tensión arterial 108/53, paraclínicos con leucocitosis de 22400 y neutrofilia de 91%, hemoglobina 15, gases arteriales con alcalemia respiratoria y Rx simple de abdomen con niveles hidroaéreos escalonados y escaso gas distal, se decidió llevar a laparotomía donde se encontró peritonitis generalizada, intususcepción de yeyuno a 60 cms del ángulo de Treitz y pólipo como lesión primaria, con perforación intestinal a ese nivel; adicionalmente, dos pólipos que ocluían la luz intestinal en íleon a 30 cms de la válvula ileocecal y en sigmoides. Se realizó resección segmentaria de yeyuno con anastomosis término-terminal, enterotomía y sigmoidotomía resecando los pólipos de esos segmentos. El informe de patología reportó en la descripción macroscópica "60 cms de intestino delgado con membranas fibrinopurulentas y áreas de necrosis segmentaria que al corte evidencia intususcepción de 50 cms con una longitud total al reducir la pieza de 120 cms y un pólipo necrótico de 9 x 5 x 1 cm con base de 1 cm. A los 15 cms se reconoció otro pólipo de 3 x 2 x 1 cms y distalmente múltiples pólipos de tamaño variable de 2 a 14 mm". Como conclusión del informe de patología "resección intestinal con necrosis difusa extensa por pólipos hamartomatosos múltiples. Masas polipoideas en íleon y colon sigmoide: pólipos hamartomatosos de Peutz-Jeghers". El paciente evoluciona satisfactoriamente dándosele salida al 6º día postoperatorio. Con el reporte de patología, se revisó en consulta externa encontrando múltiples lesiones pigmentadas en orejas, dorso nasal, región perioral, labios, mucosa oral, palmas de las manos, plantas de los pies y en región perianal. Concluyendo el diagnóstico de síndrome de Peutz-Jeghers, se solicitaron estudios de extensión y estudio genético. Endoscopia de vías digestivas altas de marzo de 2006 mostró múltiples lesiones polipoides Yamada III y IV entre 0,5 y 1 cm de diámetro en cuerpo y antro, múltiples lesiones polipoides en duodeno entre 0,4 y 0,6 cm de las cuales se tomaron biopsias cuyo reporte mostró "pólipos hamartomatosos de Peutz-Jeghers"; colonoscopia de marzo de 2006 mostró múltiples lesiones polipoides sésiles y pediculadas entre 7 mm y 3 cms desde el ciego hasta el recto, y se tomaron biopsias de las más grandes localizadas en sigmoides y ángulo esplénico cuya lectura de patología reporta "pólipos hamartomatosos de Peutz-Jeghers"; ecografía abdominal de marzo de 2006 normal; ecografía testicular de marzo de 2006 normal. El paciente se encuentra en sesiones de polipectomía por endoscopia y colonoscopia. Tiene pendiente el estudio de mutaciones del gen STK11.
Caso 2
Paciente de 21 años a quien a los 8 años le diagnosticaron síndrome de Peutz-Jeghers por manifestaciones dermatológicas. Realizan EVDA y colonoscopia sin evidencia de lesiones polipoides. A los 15 años presenta cuadro de abdomen agudo, y obstrucción intestinal por lo cual es valorada e intervenida quirúrgicamente en Hospital Universitario de la Samaritana. En febrero de 2007 ingresó a nuestra institución (Hospital Cardiovascular del niño de Cundinamarca) para control por gastroenterología, y al examen físico se documentan lesiones pigmentadas en dorso nasal, región perioral (cruzando el bermellón), labios, mucosa oral, palmas de las manos. Niega antecedentes familiares o personales de importancia; por diagnóstico de síndrome de Peutz-Jeghers se realiza estudios endoscópicos (abril de 2007) donde se evidencian múltiples lesiones polipoides Yamada II, III y IV entre 0,5 y 2,5 cm de diámetro en cuerpo y antro gástrico (figura 1) y múltiples lesiones polipoides en duodeno entre 0,5 y 1,5 cm de las cuales se tomaron biopsias y se realizó polipectomía a las de mayor tamaño. Colonoscopia con escasos pólipos en recto (estudio limitado por preparación). El reporte de patología evidenció "pólipos hamartomatosos de Peutz-Jeghers". Se decidió, en mayo de 2008 realizar enteroscopia de seguimiento evidenciando múltiples y grandes lesiones polipoides de 0,5 a 3 cms en yeyuno y parte del íleon (figura 2) que dificultaban técnicamente la progresión del enteroscopio. El abordaje rectal mostró escasas lesiones polipoides en recto y colon, que eran de gran tamaño, más de 3,5 cms que impedían la progresión del enteroscopio (figura 3). Se decide, en Junta, la realización de estudios de seguimiento y la realización de polipectomía enteroscópica en cirugía vs. enterotomías y polipectomías quirúrgicas. Tiene pendiente el estudio de mutaciones del gen STK11.
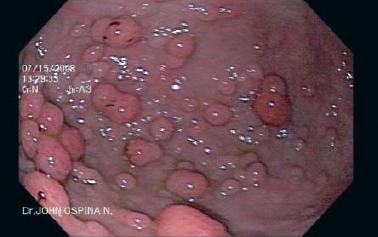
Figura 1. Pólipos gástricos.
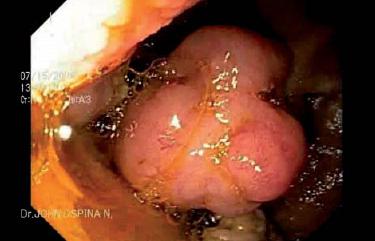
Figura 2. Pólipos en yeyuno
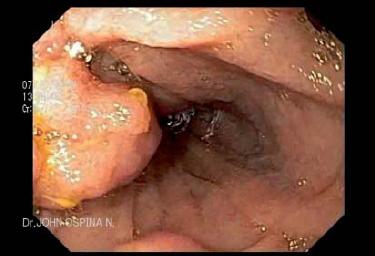
Figura 3. Pólipos en colon
Discusión
Historia
La primera descripción de esta alteración fue realizada por el doctor JRT Conner en 1895 en la Aesculapian Society of London (1, 2), en dos hermanas que padecían síndrome anémico y presentaban pigmentaciones en labios y boca, la primera de las cuales falleció a los 20 años por un cuadro de obstrucción intestinal y la segunda a los 52 años víctima de un cáncer de seno.
En 1921, el doctor Johannes Peutz (1, 3) describe la relación existente entre pigmentación mucocutánea y poliposis intestinal al estudiar a 7 miembros de una familia en 3 generaciones; posteriormente, el doctor Jeghers, en 1949 (1, 4) publicó la descripción clínica del síndrome reconociendo un carácter hereditario dominante con patrón mendeliano simple y su relación con un riesgo aumentado de cáncer. En 1954, el doctor Bruwer, de la Mayo Clinic acuñó el término síndrome de Peutz-Jeghers (1, 5) con el cual reconocemos hoy día la entidad. Sin embargo, fue solo hasta 1997, cuando los Drs. Hemminki y Amos definieron la mutación genética responsable del síndrome (1, 7-10).
Epidemiología
En Estados Unidos se ha calculado una incidencia de 1 por cada 120.000 a 200.000 nacimientos y la prevalencia se ha estimado en 1 por cada 8.300 a 29.000 nacimientos. Es igual en hombres y en mujeres, en todas las razas, y la edad promedio de diagnóstico es a los 23 años (1, 5, 6).
En Colombia no hay estadística sobre incidencia ni prevalencia y la literatura colombiana es escasa y casi siempre menciona este síndrome únicamente como factor de riesgo en artículos que tratan generalmente sobre cáncer y poliposis o complicaciones abdominales, y son, en su mayoría, publicaciones de dermatología.
Patogenia
Hasta el momento se han descrito más de 145 mutaciones relacionadas con este síndrome (1, 11), la mayoría de las cuales son pequeñas deleciones, inserciones o sustituciones simples de bases, en el gen serine/threoninie kinase (STK11) localizado en la región telomérica del brazo corto del cromosoma 19 en la banda 13,3 (19p13.3). Este gen, que se expresa en todos los tejidos humanos y con mayor intensidad en testículos y en hígado fetal, expresa una proteína (serina treonina kinasa) de 433 aminoácidos que se encuentra en el núcleo y en el citoplasma y cuya función no es completamente conocida pero que aparentemente está envuelta en el detenimiento del ciclo celular en G1 (1, 12). Esta proteína tiene que ver además en el desarrollo de la arquitectura celular, manteniendo la polaridad celular y su mutación conlleva a una pérdida de la polaridad y una tendencia al prolapso epitelial que resulta finalmente en la formación de pólipos (13-15).
Algunos investigadores, como el Dr. Karuman, han demostrado que el gen se asocia físicamente con el gen p53 regulando específicamente la vía de apoptosis (16); así como la relación existente con el factor de crecimiento endotelial vascular VEGF, (proteína que se requiere en el desarrollo normal del sistema gastrointestinal) (1, 17, 18).
La mutación del gen además codifica una proteína truncada, una proteína con plegamiento anormal, o una proteína con estructura alterada, una proteína sin sentido con dominios catalíticos incompletos que tiene como consecuencia la disminución de la actividad kinasa de la misma la cual es importante en su efecto como gen supresor tumoral (1, 8, 10, 11).
Estas mutaciones germinales del gen, asociadas a defectos genéticos adquiridos del segundo alelo en células somáticas resultan en las manifestaciones fenotípicas del síndrome (1, 19, 20-25).
Recientemente, Bardeesy y colaboradores (1, 28) sugirieron que la pérdida del gen STK11 en un epitelio da como resultado pólipos benignos y que la pérdida en una lesión en estadio tardío facilita un potencial maligno a la lesión. Al parecer, la haploinsuficiencia es suficiente en la formación de pólipos (26) y la pérdida de la heterogeneidad es necesaria en el proceso de carcinogénesis (27). Mutaciones de los genes p-53 y de β-catenina pero no en el gen APC son importantes en el proceso de carcinogénesis en estos pacientes después de la mutación del gen STK11 (30, 60).
Patrón hereditario
El 75% de los pacientes se presenta con un patrón heredo familiar autosómico dominante y el 25% es esporádico (como los casos presentados) y están asociados a mutaciones en el gen STK11 en el 60% y el 50%, respectivamente, lo que sugiere heterogeneidad genética. Los casos esporádicos son debidos a mutaciones de novo del gen STK11 o a variantes de baja penetrancia (1, 34). En los pacientes que llenan los requisitos de diagnóstico no asociados a mutaciones del STK11, se sugiere la existencia de mutaciones en otros genes dentro de los que se encuentran las alteraciones asociadas a los locus en los cromosomas 19q y 16q (1, 30, 32, 35-38).
Importancia clínica
El síndrome de Peutz-Jeghers conlleva un marcado riesgo de cáncer gastrointestinal y extragastrointestinal. Según Lim, la posibilidad de desarrollar cualquier cáncer a la edad de 65 años es de 37% el cual aumenta ligeramente a 47% cuando solo se estudian pacientes con mutaciones del gen STK11 (39); Los estudios del Dr. Boardman (40) sugieren además que el riesgo relativo para cualquier cáncer es de 9,9 con un riesgo relativo específico de 150,9 para cáncer gastrointestinal (3/4 partes de los casos antes de los 50 años) y de 20,3 para cáncer de seno y ginecológico (1, 19). Otros autores reportan que la frecuencia combinada de cáncer gastrointestinal y extragastrointestinal es de 93% a la edad de 65 años en este grupo de pacientes (41, 42). Aunque en conjunto los síndromes polipoides hamartomatosos (Peutz-Jeghers, poliposis juvenil, el síndrome de Cowden, etc.) son la décima parte de los síndromes adenomatosos y representan menos del 1% de los casos de cáncer de colon (43), su identificación temprana es primordial por su relación con cáncer en otros órganos.
Es de anotar que la transformación maligna en estos casos sigue la secuencia pólipo hamartomatoso-adenoma-adenocarcinoma (1, 20, 44-47, 60), teoría soportada con los focos de adenoma y displasia contenidos en los pólipos hamartomatosos grandes del síndrome de Peutz-Jeghers, aunque algunos autores sugieren que los carcinomas asociados al síndrome de Peutz-Jeghers siguen una vía molecular distinta a la clásica secuencia descrita (1, 29, 48, 49).
Manifestaciones clínicas
La clínica en estos pacientes es variable desde paciente asintomático con pigmentaciones de melanina mucocutáneas hasta emergencias abdominales y cáncer. Las presentaciones clínicas más comunes producida por los pólipos son la obstrucción intestinal (43%), el dolor abdominal (23%), sangre en heces (14%) y expulsión de un pólipo por el ano (8%). El resto de los casos (13%) se diagnostican por la pigmentación típica del síndrome (65).
Clásicamente los pacientes se caracterizan por las máculas melanocíticas café oscuro o café azuladas de 1 a 5 mm de diámetro bien definidas, localizadas agrupadas en la región alrededor de los orificios corporales ("peri-orificele"): boca, ojos, narinas y el ano, y es el compromiso labial-oral (típicamente cruzando el bermellón) el más frecuente de todos en un 94%; en un 66% se encuentra compromiso de las palmas de las manos y las plantas de los pies (50, 51) (figuras 4 y 5).
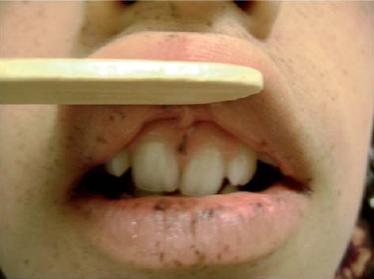
Figura 4. Compromiso labial y oral
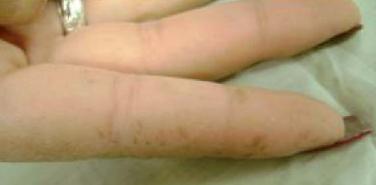
Figura 5. Depósito de melanina en manos
Estos depósitos son el reflejo de macrófagos cargados de melanina en la dermis (52) y aumento de melanocitos en la unión dermoepidérmica; se presentan en el 95% de los afectados por el síndrome, generalmente en la infancia o incluso desde el nacimiento, no tienen riesgo de transformación maligna y tienen la tendencia de ir disminuyendo en la adolescencia tardía a excepción de las localizadas en la mucosa oral (1, 4, 53-56).
La otra característica clínica predominante del síndrome es el resultado de la poliposis gastrointestinal, la cual se manifiesta a una edad temprana (57). Una tercera parte de los pacientes presentan síntomas en la primera década de la vida y hasta un 50-60% antes de la segunda década (52). Al igual que las máculas pigmentadas, se considera que la formación de nuevos pólipos disminuye con la edad (58). Los síntomas abdominales mayores son el dolor abdominal secundario a intususcepción recurrente y los cuadros de hemorragia digestiva que requieren laparotomías y resecciones intestinales múltiples con el consecuente síndrome de intestino corto. Otros síntomas incluyen dolor abdominal, y cuadros de distensión y "dispepsia". La localización más común de los pólipos es en el intestino delgado (en yeyuno más que en íleon y en este más que en duodeno) seguidos por el colon y el estómago, aunque se han descrito también en la vesícula, los pulmones, las fosas nasales y el tracto urogenital (1, 4, 41, 48).
En los casos reportados en la Clínica Mayo, el compromiso del intestino delgado fue del 96%, el colon y el recto cada uno 24% y el estómago 24% (56, 60, 61). En la serie reportada por Utsonomiya (52) el intestino delgado estaba comprometido en el 64% de los casos, el colon 64%, el estómago 49% y el recto 32%. Usualmente estos pólipos son entre uno y veinte por segmento de tracto digestivo y su tamaño varía entre 0,5 y 5 cms (62).
En una revisión de Spiegelman (65) el riesgo relativo de morir por cáncer gastrointestinal en pacientes con el síndrome es de 13 veces el riesgo de la población común, siendo tres cuartas partes de los pacientes a la edad de presentación del cáncer menores de 50 años. Esófago, estómago, intestino delgado, colon, recto, páncreas y vesícula biliar son los tumores gastrointestinales más comunes. De los tumores extragastrointestinales, cuyo riesgo relativo de presentación es de 15 veces mayor que el riesgo de la población general (38), el seno es el más común y el riesgo de cáncer es similar al encontrado en las formas hereditarias asociadas a las mutaciones de los genes BRCA-1 y BRCA-2. Otros tumores extragastrointestinales asociados al síndrome de Peutz-Jeghers son de pulmón, cuello uterino, ovario y testículo, y se presentan en estos tres últimos órganos tumores que no son muy frecuentes en la población como son el adenocarcinoma bien diferenciado de cuello uterino (adenoma maligno) (66-69), el SCTAT (sex cord tumor with annular tubules) del ovario (70-75) y el tumor de células de Sertoli del testículo (69, 73, 76).
Diagnóstico
El diagnóstico se hace en pacientes con pólipos hamartomatosos y con al menos dos de las siguientes características clínicas presentes:
- Depósitos labiales de melanina (figura 6).
- Historia familiar del síndrome.
- Pólipos del intestino delgado (figura 7).
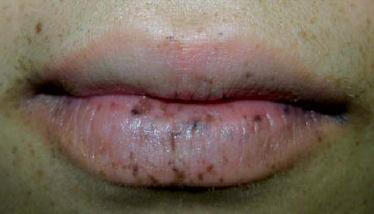
Figura 6. Depósitos labiales de melanina
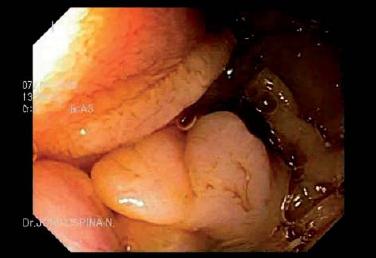
Figura 7. Pólipos en yeyuno
En ausencia de historia familiar, el diagnóstico requiere encontrar al menos dos lesiones polipoides histológicamente confirmadas como pólipos de Peutz-Jeghers (1, 38, 41).
El estudio de las mutaciones del STK11 se hace usualmente por mapeo secuencial de DNA el cual tiene una sensibilidad del 70% en familias con PJS asociadas a STK11 y una sensibilidad un poco menor en pacientes con PJS esporádico (1, 7, 11, 31). Existen estudios que sugieren que el sitio y el tipo de mutación del gen pueden influenciar el riesgo de cáncer de los pacientes, lo que apunta a que, en un futuro, la detección temprana de los portadores de mutaciones del gen podrá jugar un rol importante en el manejo y tamizaje de los mismos; incluso nuevos estudios se requerirán para demostrar si la pérdida de la heterogenicidad (LOH) en los pólipos de Peutz-Jeghers pueda ser usado como biomarcador para predecir cáncer (1, 29, 33).
Histología
Histológicamente los pólipos son hamartomas verdaderos caracterizados por el sobrecrecimiento desordenado de células nativas del órgano del que provienen, incluidas células de los tres tipos germinales en los pólipos del intestino delgado y de un solo tipo germinal en los pólipos del colon y del estómago (56, 63), con características propias que incluyen su estructura frondosa, "arboriforme", epitelio de recubrimiento específico del segmento intestinal en el que se encuentra el pólipo y un corazón central consistente en la proliferación de bandas de músculo liso de la muscularis mucosae que perforan la lámina propia y que se ramifica en cada pliegue del pólipo. Por tener este patrón arboriforme, en los cortes histológicos el epitelio puede quedar aparentemente subyacente a la lámina propia del pólipo lo que le da un aspecto invasor seudotumoral, fenómeno llamado "dislocación epitelial" y que se presenta en un 10% de los pólipos del intestino delgado mayores de 3 cms (1, 64).
Tratamiento
El manejo de la presentación clínica aguda es quirúrgico requiriendo resección de pólipos (polipectomía) o la resección del segmento intestinal comprometido. La posibilidad de procedimientos quirúrgicos repetidos existe y a largo plazo el riesgo de síndrome de intestino corto secundario a las resecciones es mayor acompañado del aumento del riesgo de muerte. Se considera que el 43% de las muertes en menores de 30 años de edad con síndrome de Peutz-Jeghers es secundario a complicaciones agudas por la poliposis más comúnmente por intususcepción y después de los 30 años hasta el 60% es atribuido a cáncer (1, 52).
Giardello y Tomlinson recomiendan polipectomía para los pólipos gástricos y del colon que sean mayores de un cm. Igualmente, se recomienda cirugía para pólipos del intestino delgado que sean sintomáticos, mayores de 1 a 1,5 cms y pólipos de crecimiento rápido (1, 49, 59, 62, 81). Algunos autores sugieren que la técnica de "clean sweep" (enterotomía y polipectomías) puede ser llevada a cabo con éxito en cirugía y aparentemente logra disminuir la necesidad de múltiples resecciones de intestino delgado (68, 69, 81). Dai y colaboradores sugieren clasificar los pacientes de acuerdo al número de pólipos por segmento, recomendando tratamiento endoscópico (enteroscopia, gastroscopia y colonoscopia) para los que tengan menos de 50 pólipos y tratamiento quirúrgico con "clean sweep" para los pacientes que tengan más de 50 pólipos (1, 60, 68, 69). Spigelman sugiere evitar al máximo resecciones intestinales y recomienda no realizar resecciones profilácticas de colon ya que el riesgo relativo de cáncer colorrectal no es tan significativo.
Es de anotar que los pólipos del intestino delgado y del colon tienden a ser pediculados y los del estómago sésiles (1, 66).
Seguimiento
El estudio y el seguimiento de los pacientes con diagnóstico de síndrome de Peutz-Jeghers se inician con endoscopia digestiva alta y una colonoscopia (71). Se le debe solicitar un estudio baritado de tránsito intestinal para valorar los pólipos del intestino delgado, o en su defecto un estudio con cápsula de endoscopia, o enteroscopia, una ecografía abdominal total con énfasis en páncreas (incluso de ser posible una ultrasonografía endoscópica); una ecografía testicular; una mamografía; una ecografía transvaginal con énfasis en anexos y biopsia endometrial; y una citología vaginal. Al caso primario, en una familia con clínica sugestiva de Peutz-Jeghers, se le solicita estudio genético para detectar mutaciones del gen STK11 (1, 48, 60, 63, 65, 68, 69, 71, 72, 81). Si estas se detectan, se debe solicitar también a todos los demás miembros de la familia en riesgo (familiares en primer grado de consanguinidad). De estos, los que tengan un resultado negativo, tienen un riesgo de tener Peutz-Jeghers igual al de la población común y por lo tanto no se benefician de ningún seguimiento; si se detecta mutación del gen, este paciente en riesgo debe entrar en un protocolo de seguimiento. En los pacientes en riesgo, incluso en las guías del John Hopkins en Baltimore, sugieren iniciar el tamizaje desde el nacimiento con historia clínica y examen físico anual buscando manchas melanóticas, pubertad precoz y tumores testiculares (1, 71). En pacientes de alto riesgo, asintomáticos y sin estigmas de Peutz-Jeghers a la edad de 8 años, se sugiere realizar la búsqueda de mutaciones del gen STK11 (39, 68, 69), argumentando que este tamizaje genético tiene la ventaja de iniciar el seguimiento a pacientes que pueden ser llevados a cirugía electiva en vez de urgencia lo cual ocurre antes de los 10 años en el 30% de los pacientes con síndrome de Peutz-Jeghers, tal como le sucedió a nuestros pacientes. Aunque no existen estudios controlados de la efectividad del seguimiento de los pacientes con síndrome de Peutz-Jeghers, una variedad de recomendaciones propuestas por expertos son expuestas en la literatura (1, 48, 65, 68, 69, 71, 72, 77, 78, 81) (tablas 1, 2 y 3).
Tabla 1. Guías de seguimiento para síndrome de Peutz-Jeghers

Tabla 2. Guías de seguimiento por año y sexo

Tabla 3. Seguimiento y recomendaciones
REFERENCIAS
1. National Center for Biotechnology Information. Online Mendelian Inheritance in Man. Available at http://ncbi.nlm.nih.gov/entrez/query.fcgi?db-OMIM. [ Links ]
2. Connor JT. Aesculapian Society of London. Lancet 1895; 2: 1169. [ Links ]
3. Peutz JLA. Very remarkable case of familial polyposis of mucous membrane of intestinal tract and nasopharynx accompanied by peculiar pigmentations of skin and mucous membrane. Nederl Maandische v Geneesk 1921; 10: 134-146. [ Links ]
4. Jeghers H, McKusick VA, Katz KH. Generalized intestinal polyposis and melanin spots of the oral mucosa, lips and digits: A syndrome of diagnostic significance. N Eng J Med 1949; 241: 993-1005. [ Links ]
5. Bruwer A, Bargen JA, Kierland RR. Surface pigmentation and generalized intestinal polyposis (Peutz-Jeghers syndrome). Proc Staff Meet Mayo Clinic 1954; 29: 168. [ Links ]
6. Mallory SB, Stough DBt. Genodermatoses with malignant potential. Dermatol Clin 1987; 5: 221-230. [ Links ]
7. Hemminki A, Tomlinson I, Markie D, et al. Localization of a susceptibility locus for Peutz-Jeghers syndrome to 19p using comparative genomic hibridization and targeted linkage analysis. Nat Genet 1997; 15: 87-90. [ Links ]
8. Hemminki A, Markie D, Tomlinson I, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature 1998; 391: 184-187. [ Links ]
9. Amos CI, Bali D, Thiel TJ, et al. Fine mapping of a genetic locus for Peutz-Jeghers syndrome on chromosome 19p. Cancer Res 1997; 57: 3653-3656. [ Links ]
10. Jenne DE, Reimann H, Nezu J, et al. Peutz-Jeghers syndrome is caused by mutations in a novel serine/threonine kinase. Nat Genet 1998; 18: 38-43. [ Links ]
11. Launonen V. Mutations update. Mutations in the human LKB1/STK11 gen. Hum Mutat 2005; 26: 291-297. [ Links ]
12. Tiainen M, Ylikorkala A, Makela TP. Growth suppression by LKB1 is mediated by a G(1) cell cycle arrest. Proc Natl Acad Sci USA 1999; 96: 9248-9251. [ Links ]
13. Martin SG, St Johnston D. A role of Drosophila LKB1 in anterior posterior axis formation and epithelial polarity. Nature 2003; 421: 379-384. [ Links ]
14. Bass AF, Kuipers J, van der Wel NN, et al. Complete polarization of single intestinal epithelial cells upon activation of LKB1 by STRAD. Cell 2004; 116: 457-466. [ Links ]
15. Jansen M, WWJ de Leng, Bass AF, et al. Mucosal prolapse in the pathogenesis of Peutz-Jeghers polyposis. Gut 2006; 55: 1-5. [ Links ]
16. Karuman P, Gozani O, Odze RD, et al. The Peutz-Jeghers gene product LKB1 is a mediator of p53-dependent cell death. Moll Cell 2001; 7: 1307-1319. [ Links ]
17. Ylikorkala A, Rossi DJ, Korsisaari N, et al. Vascular abnormalities and deregulation of VGEF in LBK1-deficient mice. Science 2001; 293; 1323-1326. [ Links ]
18. Jishage K, Nezu J, Kawase Y, et al. Role of LKB1, the causative gene of Peutz-Jeghers syndrome, in embryogenesis and polyposis. Proc Natl Acad Sci USA 2002; 99: 8903-8908. [ Links ]
19. Steinbach G, Lynch PM, Phillips RK, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Eng J Med 2000; 346: 1946-1952. [ Links ]
20. Sandler RS, Halabi S, Baron JA, et al. A randomized trial of aspirin to prevent adenomas in patients with previous colorectal carcinoma. N Eng J Med 2003; 348: 883-890. [ Links ]
21. McGarrity TJ, Peiffer L, Amos CI, et al. Overexpression of cyclooxygenase-2 in hamartomatous polyps in Peutz-Jeghers syndrome. Gastroenterology 2003; 98: 671-678. [ Links ]
22. Udd L, Katajisto P, Rossi DJ, et al. Suppression of Peutz-Jeghers polyposis by inhibition of cyclooxygenase-2. Gastroenterology 2004; 127: 1030-1037. [ Links ]
23. Seno H, Oshima M, Ishikawa TO, et al. Cyclooxygenase-2 and prostaglandin E(2) receptor EP(2) dependent angiogenesis in Apc(Delta 716) mouse intestinal polyposis. Cancer Res 2002; 62: 506-511. [ Links ]
24. Wagner TM, Mullally JF and Fitzpatrick FA. Reactive lipid species from cyclooxygenase-2 inactivate tumor suppressor LKB1/STK11: cyclopentenone prostaglandins and 4-hydroxy-2-nonenal covalently modify and inhibit the AMP- kinase that modulates cellular energy homeostasis and protein translation. J Biol Chem 2005; 281: 2598-2604. [ Links ]
25. Miyoshi H, Nakau M, Ishikawa T, et al. Gastrointestinal hamartomatous polyposis in LKB1 heterozigous knockout mice. Cancer Res 2002; 62: 2261-2266. [ Links ]
26. Esteller M, Avizienyte E, Corn PG, et al. Epigenetic inactivation of LKB1 in primary tumors associated with Peutz-Jeghers syndrome. Oncogene 2000; 19: 164-168. [ Links ]
27. Esteller M, Fraga MF, Guo M, et al. DNA mhetylation patterns in hereditary human cancers mimic sporadic tumorigenesis. Hum Mol Genet 2001; 10: 3001-3007. [ Links ]
28. Bardeesy N, Sinha M, Hezel AF, et al. Loss of the LKB1 tumor suppressor provokes intestinal polyposis but resistance to transformation. Nature 2002; 419: 162-167. [ Links ]
29. Entius M, Keller J, Westerman AM, et al. Molecular genetic alterations in hamartomatous polyps and carcinomas of patients with Peutz-Jeghers syndrome. J Clin Pathol 2001; 54: 126-131. [ Links ]
30. Mehenni H, Gehrig C, Nezu J, et al. Loss of LKB1 kinase activity in Peutz-Jeghers syndrome, and evidence for allelic and locus heterogeneity. Am J Hum Genet 1998; 63: 1641-1650. [ Links ]
31. Mehenni H, Blouin JL, Radhakrishna U, et al. Peutz-Jeghers syndrome: confirmation of linkage to chromosome 19p13.3 and identification of a potential second locus, on 19q13.4. Am J Hum Genet 1997; 61: 1327-1334. [ Links ]
32. Scott RJ, Crooks R, Meldrum CJ, et al. Mutation analysis of the STK11/LKB1 gen and clinical characteristics of an Australian series of Peutz-Jeghers syndrome patients. Clin Gen 2002; 62: 282-287. [ Links ]
33. Schumacher V, Vogel, Leube B, et al. STK11 genotyping and cancer risk in Peutz-Jeghers syndrome. J Med Genet 2005; 42: 428-435. [ Links ]
34. Giardello FM, et al. Very high risk of cancer in familial Peutz-Jeghers syndrome. Gastroenterology 2000; 119: 1447-1453. [ Links ]
35. Olschwang S, Markie D, Seal S, et al. Peutz-Jeghers disease: most, but not all, families are compatible with linkage to 19p13.3. J Med Genet 1998; 35: 42-44. [ Links ]
36. Jiang C-Y, Esufali S, Berk T, et al. STK11/LKB1 germline mutations are not identified in most Peutz-Jeghers syndrome patients. Clinical Genet 1999; 56: 136-141. [ Links ]
37. Boardman L, Couch F, Burgart L, et al. Genetic heterogeneity in Peutz-Jeghers syndrome. Hum Mutat 2000; 16: 23-30. [ Links ]
38. Tomlinson IP, Olschwang S, Abelovitch D, et al. Testing candidate loci on chromosome 1 and 6 for genetic linkage to Peutz-Jeghers disease. Ann Hum Genet 1996; 60: 377-384. [ Links ]
39. Lim W, et al. Further observations on LKB1/STK11 status and cancer risk in Peutz-Jeghers syndrome. Br J Cancer 2003; 89: 308-313. [ Links ]
40. Boardman L, Thibodeau S, Schaid D, et al. Increased risk for cancer in patients with the Peutz-Jeghers syndrome. Ann Intern Med 1998; 128: 896-989. [ Links ]
41. Tomlinson I, Houston RS. Peutz-Jeghers syndrome. J Med Genet 1997; 34: 1007-1011. [ Links ]
42. Giardello F, Welsh S, Hamilton S, et al. Increased risk of cancer in Peutz-Jeghers syndrome. N Eng J Med 1987; 316: 1511-1514. [ Links ]
43. Nagy R, Sweet K, Eng C. Highly penetrant hereditary cancer syndromes. Oncogene 2004; 23: 6445-6470. [ Links ]
44. Hemminki A. The molecular basis and clinical aspects of Peutz-Jeghers syndrome. Cell Mol Life Sci 1999; 55: 735-750. [ Links ]
45. Wang ZJ, Ellis I, Zauber P, et al. Allelic imbalance at the LKB1 (STK 11) locus in tumors from patients with Peutz-Jeghers syndrome provides evidence for a hamartoma - (adenoma) - carcinoma sequence. J Pathol 1999; 188: 9-13. [ Links ]
46. Foley T, McGarrity T, Abt and A.B. Peutz-Jeghers syndrome: a clinicopathologic survey of the "Harrisburg family" with a 49 years follow up. Gastroenterology 1988; 95: 1535-1540. [ Links ]
47. Nakayama H, Fujii M, Kimura A, et al. A solitary Peutz-Jeghers type hamartomatous polyp of the rectum: report of a case and review of the literature. Jpn J Clin Oncol 1996; 26: 273-276. [ Links ]
48. McGarrity T, Kulin H, Zaino R. Peutz-Jeghers syndrome. Am J Gastroenterol 2000; 95: 596-604. [ Links ]
49. Oncel M, Remzi M, Church J, et al. Course and follow-up of solitary Peutz-Jeghers polyps. A case series. Int J Colorectal Dis 2003; 18: 33-35. [ Links ]
50. Touraine A, Couder F. Lentiginose peri-orificielle et polypose viscerale. Presse Med 1946; 54: 405. [ Links ]
51. Traboulsi E, Maumenee J. Periocular pigmentation in the Peutz-Jeghers syndrome. Am J Ophtalmol 1986; 15: 126-127. [ Links ]
52. Utsunomiya J, Gocho H, Miyanaga T, et al. Peutz-Jeghers syndrome: its natural course and management. The John Hopkins Med J 1975; 136: 71-82. [ Links ]
53. Butterworth T, Ladda R, eds. Clinical Gastroenterology, Vol 1. New York: Praeger, 1981. p. 59-63. [ Links ]
54. Kyle J, Peutz-Jeghers syndrome. Scot Med J 1961; 6: 361-367. [ Links ]
55. Dormandy T. Gastrointestinal polyposis with mucocutaneous pigmentation (Peutz-Jeghers syndrome). N Eng J Med 1957; 256: 1186-1190. [ Links ]
56. Bartholomew L, Dahlin D, Waugh J. Intestinal polyposis associated with mucocutaneous melanine pigmentation (Peutz-Jeghers syndrome). Review of the literature and report of six cases with special reference to pathologic findings. Gastroenterology 1957; 32: 434-451. [ Links ]
57. Sommerhaug R, Monson T. Peutz-Jeghers syndrome and ureteral polyposis. JAMA 1970; 211: 120-122. [ Links ]
58. Bartholomew L, Moore C, Dahlin D, et al. Intestinal polyposis with mucocutaneous pigmentation. Surg Gynecol Obstet 1962; 115: 1-11. [ Links ]
59. Giardello F. Gastrointestinal polyposis syndromes and hereditary nonpolyposis colorectal cancer. En: Rugsti AK, ed. Gastrointestinal cancers: biology and therapy. Philadelphia Lippincott-Raven Publishers. 1995. p. 370-371. [ Links ]
60. McGarrity T, Amos C. Peutz-Jeghers syndrome: clinicopathology and molecular alterations. Cell Mol Life Sci 2006; 63: 2135-2144. [ Links ]
61. Shepherd N, Bussey H, Jass J. Epithelial misplacement in Peutz-Jeghers polyps. Am J Surg Pathol 1987; 11: 743-749. [ Links ]
62. Giardello F, Trimbath J. Peutz-Jeghers syndrome and management recommendations. Clin Gastroenterol Hepat 2006; 4: 408-415. [ Links ]
63. Spigelman A, Murday V, Phillips R. Cancer and Peutz-Jeghers syndrome. Gut 1989; 30: 1588-1590. [ Links ]
64. Chen K, Female genital tract tumors in Peutz-Jeghers syndrome. Hum Pathol 1986; 17: 858-861. [ Links ]
65. Spigelman A, Arese P, Phillips R. Polyposis: the Peutz-Jeghers syndrome. Br J Surg 1995; 82: 1311-1314. [ Links ]
66. Oncel M, Remzi F, Church J, et al. Benefits of "clean sweep" in Peutz-Jeghers patients. Colorectal Dis 2004; 6: 332-335. [ Links ]
67. Dai Y, Song Y, Xiao B, et al. Clinical classification of Peutz-Jeghers syndrome. J South Med Univ 2006; 26: 79-81. [ Links ]
68. Guillem J, Smith A, Puig-La Calle J, et al. Gastrointestinal polyposis syndrome. Curr Probl Surg 1999; 30: 291-294. [ Links ]
69. Williams C, Goldblatt M, Delaney P. "Top and tail endoscopy" and follow-up in Peutz-Jeghers syndrome. Endoscopy 1982; 14: 82-84. [ Links ]
70. Grady W. Genetic testing for high-risk colon cancer patients. Gastroenterology 2003; 124: 1574-1594. [ Links ]
71. The Johns Hopkins guide for patients and families: Peutz-Jeghers syndrome. Baltimore: Johns Hopkins University, 2001. [ Links ]
72. Burke W, Daly M, Garber J, et al. Recommendations for follow-up care of individuals with an inherited predisposition to cancer. II. BRCA1 and BRCA2: Cancer Genetics Studies Consortium, JAMA 1997; 227: 997-1003. [ Links ]
73. Genetic/familial high-risk assessment: breast and ovarian: National Comprehensive Cancer Network. Clinical practice guidelines in oncology- v.1.2004. Available at; http://www.nccn.org/professionals/ physicians_gls/PDF/genetics_screening.pdf. [ Links ]
74. Hereditary breast and ovarian cancer syndromes. In: Offit K, Garber J, Grady W, et al. eds. ASCO curriculum: Cancer genetics & cancer predisposition testing. 2nd ed. Alexandria, VA: ASCO Publishing, 2004. p. 6.1-6.55. [ Links ]
75. Dunlop M. Guidance on gastrointestinal surveillance for hereditary nonpolyposis colorectal cancer, familial adenomatous polyposis, juvenile polyposis, and Peutz-Jeghers syndrome. Gut 2002; 51(suppl): V21-V27. [ Links ]
76. Colorectal cancer screening: National Comprehensive Cancer Network. Clinical practice guidelines in oncology- v.1.2004. Available at; http://www.nccn.org/professionals/physicians_gls/PDF/colorectal_ screening.pdf [ Links ]
77. Brentnall T, Bronner M, Byrd D, et al. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med 1999; 131: 247-255. [ Links ]
78. Canto M, Goggins M, Yeo C, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol 2004; 7: 606-621. [ Links ]
79. Parsi M, Burke C. Utility of capsule endoscopy in Peutz-Jeghers syndrome. Gastrointest Endosc Clin N Am 2004; 14: 159-167. [ Links ]
80. Giardello F, Brensinger J, Petersen G. AGA technical review on hereditary colorectal cancer and genetic testing. Gastroenterology 2001; 121: 198-213. [ Links ]
81. Finding cancer early: ACS guidelines: American Cancer Society. Available at: http://www.cancer.org. [ Links ]

