










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.24 no.3 Bogotá July/Sept. 2009
Carcinogénesis gástrica
Gastric carcinogenesis
William Otero Regino, MD (1), Martín A. Gómez MD (2), Denny Castro MD (3)
(1) Profesor Asociado de Medicina, Unidad de Gastroenterología Universidad Nacional de Colombia, Gastroenterólogo de Clínica Fundadores, de Fundación Hospital San Carlos y de la Clínica Carlos Lleras Restrepo, Bogotá, Colombia.
(2) Profesor de Medicina Unidad de Gastroenterología, Universidad Nacional de Colombia, Gastroenterologo de Hospital El Tunal, Fundacion Hospital San Carlos y Clínica Carlos Lleras Restrepo, Bogotá, Colombia.
(3) Director General y Director de Posgrado de Gastroenterología, del Centro de Control de Cáncer Gastrointestinal Doctor Luis E Anderson San Cristobal, Estado Táchira Venezuela.
Fecha recibido: 02-02-09 Fecha aceptado: 12-08-09
Resumen
El cáncer gástrico (CG) es la segunda causa de muerte por cáncer. Más del 90% de los CG son adenocarcinomas y el principal agente etiológico es H. pylori y aunque este es necesario, no es suficiente ya que solo 1-2% de los infectados desarrolla CG. Su origen es multifactorial, e involucra factores genéticos del individuo, factores medioambientales y la infección por H. pylori. Los mecanismos por los cuales H. pylori participa en la carcinogénesis no son claros pero hay dos vías involucradas: mecanismos indirectos a través de la inflamación persistente inducida por la infección, acompañada de hiperproliferación celular, y daño del DNA por radicales libres, con participación adicional de células progenitoras de la médula ósea que sería el "stem cell" para el CG. La segunda vía involucra acciones directas de proteínas de H. pylori sobre las células gástricas. Entre los factores genéticos del huésped hay evidencia de que polimorfismos genéticos de IL-1B, TNF, IL-8, INF gama e IL-10 entre otros, inducen una fuerte respuesta inflamatoria que se asocia con mayor riesgo de CG.
Palabras clave
Helicobacter pylori, carcinogénesis, células progenitoras, polimorfismos.
Summary
Gastric cancer is second among cancers as a cause of death. More than 90% of all gastric cancers are adenocarcinomas whose principal cause is Helicobacter pylori. Although H. Pylori is a necessary condition, it is not a sufficient condition since only 1-2% of those infected develop gastric cancer. There are multiple factors besides H. Pylori infection involved in the etiology of this cancer. They include genetic factors related to the individual and environmental factors. Although the ways in which H. Pylori participates in this carcinogenesis are not completely clear, two different mechanisms are involved. H. Pylori infection induces persistent inflammation accompanied by hyperproliferation of cells, and it causes damage to DNA from free radicals in which progenitor cells from the bone marrow participate. These cells could be the "stem cells" of gastric cancer. The second path involves the direct action of proteins from H. Pylori on gastric cells. Among the genetic factors involved there is evidence that IL-1B, TNF, IL-8, and INF gama e IL-10 polymorphisms, among others, induce B inflammatory responses which are associated with higher risks of gastric cancer.
Key words
Helicobacter pylori, carcinogenesis, stem cell, polymorphisms.
El cáncer gástrico (CG) es una entidad heterogénea y altamente prevalente en el mundo. En el 2002 se estimó que hubo 900.000 casos nuevos y 700.000 muertes (1). Globalmente, es el cuarto tipo de cáncer más frecuente y la segunda causa de muerte por cáncer (2), explicando el 10% de las mismas (3), aunque en Japón (4) y en Colombia (5) es la primera causa de muerte por cáncer. En este último país su incidencia es aproximadamente 10 veces más alta que en USA (5).
Más del 90% de los CGs son adenocarcinomas (6, 7) y el resto tumores menos frecuentes como linfomas, tumores estromales gastrointestinales (GIST) y tumores carcinoides (7). El principal agente etiológico de los CG distales o no cardiales, y de los linfomas tipo MALT es Helicobacter pylori (H. pylori) (8). En las personas infectadas el CG se desarrolla en 1-2% y el linfoma MALT de bajo grado de malignidad, en 0,7 a 0,8 por 100.000 individuos, aunque en USA se ha informado de 1 caso entre 30.000-80.000 individuos y en Italia hasta 13 casos por 100.000 individuos (9).
De acuerdo a la clasificación de los patólogos finlandeses Jarvi y Lauren, los CGs se dividen en dos tipos histológicos: intestinal y difuso (10, 11), los cuales tienen claras diferencias desde el punto de vista epidemiológico, histopatológico, endoscópico, clínico y patogenético (12).
La mayoría de los CGs son esporádicos, pero aproximadamente el 10% tiene agrupación familiar y del 10 al 30% de estos son hereditarios (13). Los de tipo intestinal son los más frecuentes en los sitios de alta prevalencia de CG, tienen mejor pronóstico y ocurren más a menudo en hombres, a partir de los 50 años, aunque en sitios de alta prevalencia aparece a menor edad (6,7). En sitios de baja prevalencia o de bajo riesgo, como en muchos países desarrollados, su incidencia ha disminuido en las últimas décadas (7), Este tipo de CG aparece en estómagos que tienen gastritis atrófica y metaplasia intestinal y el mayor número de casos ocurre en países subdesarrollados (6, 7, 14).
El tipo difuso, por el contrario, no muestra variación geográfica, es más frecuente en mujeres, aparece en personas jóvenes, con frecuencia hay historia familiar positiva, tiene peor pronóstico y no se acompaña de atrofia gástrica ni metaplasia intestinal (6, 14); su incidencia no ha variado e incluso parece estar aumentando (15, 16). La endoscopia digestiva alta, que es el mejor método para el diagnóstico de CG, con frecuencia tiene dificultades para el diagnóstico del CG difuso, ya que este no tiende a formar masas exofíticas sino que sigue un patrón de infiltración submucosa (16, 17). El diagnóstico es aún más difícil cuando hay linitis plástica.
El 1-3% de todos los CGs son hereditarios y este subgrupo representa un síndrome clínico-patológico claramente definido, conocido como el síndrome de CG difuso hereditario (17, 18). En el 30% de las familias con esta entidad, se han encontrado mutaciones en el gen epitelial de caderina (E-cadherin) (CDH1), una proteína de adhesión celular (19-22). La mutación es autosómica dominante con penetrancia del 70%, es decir, quien la padece tiene un riesgo de 70% de padecer CG y las mujeres mayor riesgo adicional de padecer cáncer de seno, variedad lobulillar (19, 22). El CGH tiene alta mortalidad a edad temprana y cuando se diagnostica, la mayoría de pacientes ya tiene enfermedad avanzada y alto riesgo de recaída postratamiento quirúrgico (16). En los individuos con la mutación CDH1, se recomienda gastrectomía profiláctica y en quienes no la aceptan, endoscopia digestiva alta cada 6-12 meses (22). Hoy día, se recomienda investigar las mutaciones para CDH1 en las siguientes situaciones (22):
-
Familias con dos o más casos de CG difuso, uno de los cuales diagnosticado antes de los 50 años de edad.
-
Familias con más de tres casos de CG difuso, diagnosticados a cualquier edad.
-
Individuos aislados con diagnóstico de CG difuso antes de 35 años.
-
Individuos aislados con cáncer de seno tipo lobulillar y CG difuso.
-
Familias con un miembro con CG difuso y otro con cáncer de seno o cáncer de colon con células en anillo de sello. También se recomienda la prueba para familias con múltiples casos de cáncer de seno lobulillar con o sin un caso de CG difuso, ya que las mutaciones CDH1, también se han detectado en familias con solamente cáncer de seno de tipo lobulillar (23).
Teniendo en cuenta que la mayoría de los casos de CG son de tipo intestinal, este artículo se centrará en las vías carcinogénicas involucradas en el mismo y no se tratarán los otros tumores. La inspiración para realizar esta revisión fueron los doctores Pelayo Correa y David Graham, dos connotadas figuras universales, que han contribuido de manera sustancial en el conocimiento de esta importante patología.
El CG es una enfermedad multifactorial que se desarrolla a través de un proceso de múltiples pasos que puede durar 20 años o más (24). Su génesis es compleja e involucra la participación de tres factores principales: factores medioambientales, el agente (H. pylori) y los factores genéticos del huésped (6, 7, 12, 14, 24, 25).
Medioambiente
Dieta: múltiples estudios observacionales han examinado la asociación entre el consumo de frutas y vegetales y el riesgo de CG. Algunos han encontrando que las frutas frescas, vitamina C y betacarotenos, se asocian a menor riego de CG (26-29) aunque en otros no encontraron este efecto (30). En una revisión de Cochrane, se concluyó que no hay evidencia de que los suplementos dietarios con antioxidantes, incluyendo vitamina C, reducen el riesgo de CG (31). Sin embargo, no hay ensayos clínicos que hayan estudiado la eficacia del consumo de vegetales y frutas sobre el riesgo de CG y la evidencia de su efecto protector proviene solamente de estudios epidemiológicos (32).
Los resultados de ensayos clínicos con betacarotenos y otros antioxidantes para prevenir CG no son consistentes. En Colombia, Correa y col (33) encontraron que un suplemento de 6 mg de betacarotenos diariamente durante seis años, aumentó significativamente la tasa de regresión de la atrofia gástrica y de la metaplasia intestinal. Sin embargo, el seguimiento a largo plazo de los pacientes de este ensayo encontró que el beneficio de la vitamina C y de los betacarotenos desapareció con el tiempo, al contrario de lo que sucedió al erradicar H. pylori (34). Estos resultados, se correlacionan con el trabajo de Blot y col, de la China, en el cual encontraron que suplementos con betacaroteno (15 mg), alfa tocoferol (30 mg) y selenio 50 ugr durante 5 años, no modificó la mortalidad por CG del cardias ni de los distales o no cardiales (35).
Experimentalmente, los carotenoides (Licopene, lutein y betacarotenos) y los retinoides, inhiben el crecimiento de tumores gástricos químicamente inducidos en animales de laboratorio (36-39). Se cree que el beneficio de los carotenoides en génesis del CG, se deriva de diversos mecanismos tales como: disminución de la proliferación celular y al mismo tiempo, efecto antioxidante que bloquea radicales libres evitando el daño oxidativo del DNA (37, 39) e inducción de apoptosis (40), disminución la población bacteriana de H. pylori y reducción de la inflamación al corregir la respuesta inmune hacia Th2, en vez de la respuesta equivocada que paradójicamente es Th1 (41, 42).
Sal: hay evidencia de mayor riesgo de CG en los individuos que tienen alta ingesta de sal o un alto consumo de alimentos preservados en sal (7, 9). En ocho estudios de la Fundación para la investigación de cáncer en el mundo (WCRF) y el Instituto americano para la investigación en cáncer (AIRC) (43), se encontró aumento del riesgo de CG (OR 2,1 a 5,0) con el consumo de sal pero en cuatro no se encontró asociación. Experimentalmente, la sal aumenta los tumores gástricos (44, 45). Altas concentraciones de sal en el estómago producen diversos efectos dañinos sobre el mismo: inflamación, daño de la capa de moco, aumento de la proliferación celular y síntesis del DNA, los cuales pueden aumentar el riesgo de CG en un medioambiente de inflamación constante (46, 47). También se ha demostrado que el daño mucoso inducido por la sal aumenta la persistencia de la infección por H. pylori en ratones (48).
Nitrato, nitritos y nitrosaminas
Los seres humanos se exponen a dos fuentes de nitrosaminas. La primera son nitrosaminas preformadas, presentes en carnes, pescados y otros alimentos preservados con nitritos, encurtidos ahumados, alimentos salados y bebidas alcohólicas como cervezas y whiskys (49). La segunda fuente son los nitratos de los vegetales utilizados como aditivos en quesos y carnes curadas (50). Los nitratos de la dieta pueden ser reducidos a nitritos por las bacterias de la cavidad oral y estos a compuestos N-nitrosos por bacterias presentes en el estómago por reacciones con amidas, aminoácidos y aminas (50) o también por la formación de óxido nítrico cuando hay inflamación (32, 49, 50). Se ha encontrado mayor riesgo de CG con la formación de nitrosaminas cuando hay infección por H. pylori o disminución de los niveles plasmáticos de vitamina C (51). Diferentes estudios experimentales y observacionales sugieren que la nitrosamina y el consumo de alimentos procesados con sustancias relacionadas con aquella, aumentan el riesgo de CG (32).
Alcohol: su asociación con CG es controvertida y los datos disponibles no apoyan el concepto de que esta sustancia se asocie con mayor riesgo de CG (32).
Helicobacter pylori
H. pylori fue el primer patógeno bacteriano en ser clasificado como carcinógeno tipo I por la Agencia internacional para investigación en cáncer (52). Esta calificación, se basó en los resultados de tres grandes estudios epidemiológicos (53-55), antes de que la asociación se hubiera demostrado experimentalmente (56, 57). Este pronunciamiento de la IARC, sin evidencia experimental, fue criticado por algunos científicos, por considerarlo apresurado. Los estudios epidemiológicos mencionados demostraron que el riesgo de CG en los infectados estaba entre 2,8 y 6 veces al compararlos con los no infectados. El estudio internacional EUROGAST encontró también que los infectados tenían un mayor riesgo de padecer CG, con un OR de 6 (58) y en un metanálisis relativamente reciente, el riesgo para CG fue de 2,28 para los individuos con serología positiva para H. pylori y 2,87 cuando el microorganismo era CagA (+) (59). Los estudios mencionados y otros más recientes, han permitido concluir que la evidencia sobre la relación ente H. pylori y CG es inequívoca y que este microorganismo es el principal agente causal de este tumor (60-63); sin embargo, una pequeña proporción de CGs, posiblemente está asociada con el virus de Epstein Barr(64).
Actualmente se considera que H. pylori es el responsable de más del 90% de los CGs (62), aunque no es un factor suficiente ya que solo una minoría de los infectados, tendrán CG. Hoy día ya no se discute la asociación causal de esta infección, sino los mecanismos por los cuales se genera el CG y cómo se identifica a los individuos con mayor riesgo.
Todos los H. pylori son patógenos y producen gastritis crónicas en todas las personas infectadas (62, 65-68). Sin embargo, unos microorganismos son más virulentos que otros. La gastritis crónica es asintomática y su patrón difiere según el desenlace clínico final: gastritis antral en los casos de úlcera duodenal y pangastritis/gastritis corporal en casos de cáncer gástrico o úlcera gástrica (68-71) (figura 1). La asociación entre estos dos tipos de gastritis con sus respectivas enfermedades ha sido reconocida desde hace más de 70 años (72, 73).
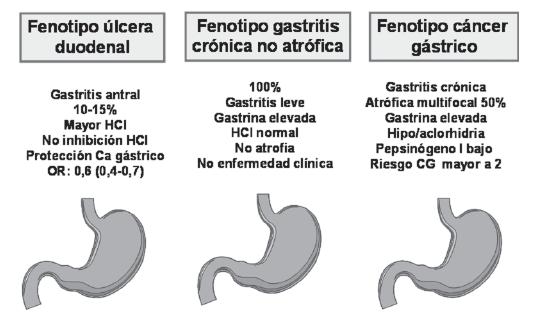
Figura 1. Patrones de gastritis asociados a H. pylori. Modificado de ref. 108. Amieva MR, El-Omar. Gastroenterology 2008; 134: 306-23.
Las consecuencias finales de la infección dependen de genéticos del huésped, características de H. pylori y factores medioambientales externos. Los dos tipos de gastritis crónica producen diferentes alteraciones de la fisiología gástrica. En la gastritis antral hay hipersecreción de ácido (70, 71), el cual resulta en metaplasia gástrica del bulbo duodenal con alteración de sus mecanismos de defensa y finalmente úlcera duodenal. Desde hace mucho tiempo se sabe que los pacientes con úlcera duodenal (UD), a pesar de tener gastritis antral, tienen menor riesgo de CG (62, 69, 74), mientras que la úlcera gástrica aumenta el riesgo para CG (75). En Japón, donde el CG tiene alta incidencia y prevalencia, la relación UD/UG es menor de 1 (76) y en regiones de baja prevalencia de CG como algunos países occidentales y el sureste asiático, esa relación es mayor de 1 (77, 78). En Japón, Uemura y col (74) encontraron que ninguno de los pacientes con UD infectados por H. pylori tuvieron CG durante un seguimiento de ocho años en contraste con 3,4% de los pacientes con UG. En ese estudio también se corroboró que los pacientes con UD generalmente tenían gastritis antral con poca o ninguna atrofia, mientras que los que tenían UG, generalmente tuvieron gastritis corporoantral con grados variables de atrofia. La gastritis predominantemente corporal, se asoció con un riesgo relativo (RR) para CG de 34,5 (74).
Los mecanismos por los cuales H. pylori produce CG no se conocen completamente, pero en la actualidad se considera que la carcinogénesis puede involucrar mecanismos indirectos (inflamación permanente) y mecanismos directos representados por la acción de los diferentes factores de virulencia de H. pylori sobre el epitelio gástrico (76).
Mecanismos indirectos
Estos se relacionan con la fuerte respuesta inflamatoria producida en el estómago infectado, la cual causa cambios moleculares y morfológicos en el epitelio originando la siguiente secuencia histopatológica: gastritis crónica, atrofia gástrica, metaplasia intestinal completa, metaplasia intestinal incompleta, displasia y cáncer en 1-2% de los infectados (68, 79, 80). La atrofia, que usualmente está presente, puede ser multifocal o tener un patrón difuso y asociada a una forma de metaplasia mucosa, denominada seudopilórica ("antralización del cuerpo") (81), también conocida como metaplasia que expresa el polipéptido espasmolítico, ya que estas glándulas con morfología antral presentes en el cuerpo gástrico, expresan este polipéptido, que es un péptido trefoil, normalmente presente en la mucosa intestinal normal y en células displásicas y neoplásicas (82).
Estas vías hipotéticas sugieren que la inflamación crónica lleva a atrofia gástrica, la cual sería una lesión preneoplásica gástrica. La inflamación crónica produce un aumento del recambio tisular, con excesiva proliferación celular la cual puede predisponer a errores mitóticos más frecuentes con mayor riesgo de mutagénesis (69, 76, 80, 82). La concurrencia de hiperproliferación celular con inflamación que involucra la generación citoquinas, factores de crecimiento y de radicales libres de oxígeno y de nitrógeno (óxido nítrico) (62, 68, 83), favorecen la posibilidad de daño del DNA de las células gástricas, el cual puede inducir mutaciones en el DNA o "silenciar" genes a nivel de su trascripción (62, 68). Matsumoto y col encontraron que la infección por H. pylori produjo expresión de la activación inducida por el gen deaminasa citidina (AID) (84), que puede predisponer a mutaciones puntuales en el gen supresor de tumores p53 (85). Factores lesivos del medio ambiente, tales como el tabaquismo y altos contenidos de sal en la dieta, aumentan adicionalmente el riesgo de CG, mientras que dietas con altos contenidos de antioxidantes como las verduras y frutas frescas pueden ser protectoras (31).
Esta secuencia de eventos constituye el paradigma clásico del modelo del profesor Pelayo Correa (12, 26, 68). El doctor Correa tiene el mérito de haber propuesto la teoría sobre la carcinogénesis gástrica, hace más de 30 años, antes del descubrimiento de H. pylori, como un proceso de múltiples pasos que involucra la progresión de gastritis a cáncer (86). El paradigma del doctor Correa, progresivamente se hizo más relevante con el descubrimiento de H. pylori como el principal causante de gastritis crónica (87) (figura 2). Este modelo se ha ido modificando cada vez más, a medida que se han establecido diferentes alteraciones que influyen en la historia natural de la infección (82). Correa identificó que la gastritis atrófica era multifocal, ocurría en todo el estómago y era más frecuente en las áreas geográficas con mayor incidencia de CG (88, 89). Además, demostró que las poblaciones con mayor riesgo de CG en Colombia tenían mayor prevalencia de gastritis atrófica que aquellas con menor riesgo para el tumor, comprobando observaciones hechas previamente por otros autores (90, 91). Aunque en su modelo de carcinogénesis, el doctor Correa considera que el CG se inicia con gastritis crónica, y esta más tarde progresa hacia atrofia y luego a metaplasia intestinal-displasia-cáncer, de una manera secuencial, hasta el momento no se sabe si realmente la atrofia precede a la metaplasia o si por el contrario, ambas ocurren simultáneamente. La verificación de esta secuencia solo sería posible si las lesiones individuales se pudieran seguir prospectivamente sin ninguna intervención (92).
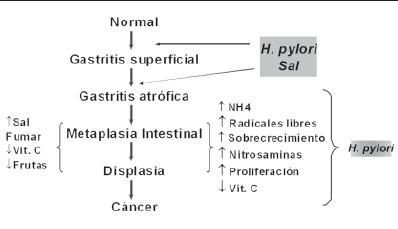
Figura 2. Carcinogénesis, modelo de Pelayo Correa.
El concepto de la asociación entre inflamación y CG ya había sido reconocido por Virchow en 1863, cuando hipotetizó que el cáncer se origina en los sitios con inflamación crónica (93).
La infección por H. pylori estimula tanto la respuesta inmune innata como la adquirida (94). El paso inicial en este proceso es el reconocimiento del microorganismo a través de Nod1 ("nucleotide-binding oligomerization domain protein I"), que es un sistema innato de detección de bacterias gram negativas que identifica un neuropéptido de los peptiglicanes de estas bacterias (94-97).
La estimulación del sistema inmune después del reconocimiento de H. pylori por el Nod1, resulta en gastritis crónica por las células inflamatorias que infiltran el epitelio colonizado, las cuales tendrán influencia en la densidad de la colonización, el nivel de inflamación y la generación de la respuesta inmune adaptativa (94, 95). De esta manera, la respuesta innata es un determinante fundamental de la severidad de la enfermedad y de la carcinogénesis gástrica. Esta respuesta inmune innata contra H. pylori incluye la liberación de péptidos antibacterianos y de infiltración de la mucosa por todos los tipos de células efectoras inmunológicas. Un inadecuado reconocimiento de H. pylori por el sistema inmune innato puede contribuir a la falla del sistema inmune adaptativo para eliminarlo. Determinados polimorfismos del receptor "Toll-like 4" (TLR4+3725 G/C) se han encontrado asociados con mayor riesgo de atrofia gástrica (99) y de CG (100), indicando que esta variación de proteínas transmembrana de reconocimiento de patrones moleculares de patógenos del sistema inmune innato son factores del huésped que participan en la respuesta y desenlace de la infección.
Además de la respuesta primaria, también se produce una respuesta inmunológica adquirida celular y humoral, local y sistémica, que persiste durante toda la vida. La respuesta de la célula T es fundamentalmente Th1 (96-98, 101, 102), una respuesta "equivocada", ya que H. pylori es un germen extracelular, que al igual que microorganismos similares, debería desencadenar una respuesta Th2 (96, 97, 101, 102). La respuesta Th1 produce interferón gama (IFN g) factor de necrosis tumoral alfa (TNF a). IL-12, IL-18 (94-97). La polarización de la respuesta inmune hacia Th1, con su respectivo perfil de citoquinas, puede contribuir al desarrollo de una patología gástrica más severa y por el contrario, la activación de una respuesta Th2 y la expresión de sus citoquinas como IL-4, produce disminución de la inflamación gástrica y protección contra patologías más severas, incluyendo probablemente el CG, al contrarrestar los efectos de las citoquinas Th1(96, 101, 102). Estas citoquinas tipo I activan los macrófagos, los cuales a su vez secretan factores proinflamatorios y adquieren mayor capacidad bactericida en comparación a su activación por respuesta celular Th2 (95-97). La severidad de la gastritis crónica, se correlaciona con el número de células que secretan IFN g (96, 62, 101, 102). La diferenciación de la respuesta inmune hacia Th1 parece que es influida por la bacteria misma y por factores medioambientales (62, 68, 96, 101). Las infecciones por parásitos, que inducen respuesta Th2 se ha sugerido como uno de los factores que explica la menor incidencia de cáncer gástrico en regiones del África con alta prevalencia de infección por H. pylori ("enigma africano") (103), aunque este "enigma" ha sido desafiado por algunos expertos, quienes consideran que realmente es un mito, por cuanto en ese continente realmente si hay úlceras duodenales y CG (73). Recientemente se ha encontrado que la IL-17 y la IL-23, están involucradas en la infección por H. pylori (96). La IL-17, citoquina, producida por el linfocito Th17 altera la vigilancia inmune y promueve el crecimiento tumoral (96). En China, Zhang y col (104) encontraron que la IL-23 y la IL-17 estaban significativamente elevadas en pacientes con CG avanzado, sugiriendo que el linfocito Th17 puede contribuir en la carcinogénesis promovida por H. pylori.
Cuando una persona está infectada por H. pylori, el riesgo de CG es de 2 a 3 veces más, pero si tiene anticuerpos anti-CagA el riesgo aumenta a 11 y dependiendo del grado de fosforilación de la proteína puede aumentar aún más (105, 106). Si a todo lo anterior, se suma la alteración del gen que codifica la síntesis de la IL-1B-511, el riesgo aumenta a 87 veces (68) (tabla 1). La modulación del proceso inflamatorio, en gran parte determina el desenlace neoplásico o no (60, 62, 68, 79, 70). Una gran diferencia entre estas dos posibilidades es la secreción de ácido que, como se mencionó, los pacientes con UD la tienen normal o aumentada y los pacientes con úlcera gástrica y gastritis corporoantral, generalmente tienen hiposecreción de ácido (62, 67, 70, 79). En este último tipo de gastritis, los factores del huésped como IL-1, IL-8 y metaloproteinasas de la matriz y factores de virulencia de H. pylori como las proteínas CagA, VacA y OipA, juegan un papel importante en el desarrollo de la úlcera gástrica (60, 107-110).
Tabla 1. H. pylori y riesgo de cáncer gástrico.

La hiposecreción gástrica es, en parte, determinada por la interleukina 1B (IL-1B), la cual, además de ser una citoquina proinflamatoria, es un potente inhibidor de la secreción de ácido (100 veces más potente que el omeprazol) (111). Algunos han encontrado que polimorfismos del gen de la IL-1B y del gen del antagonista de la IL-1 (IL-1RN) no solo se asocian con hipoclorhidria sino también con mayor riesgo CG (112-114), aunque otros no han encontrado la asociación con CG (115-117). Sobre esta controversia se han realizado tres metanálisis incluyendo las múltiples publicaciones sobre el tema y analizando el polimorfismo IL-1B y el IL-1RN (118-120). Dos de ellos concluyeron que el genotipo proinflamatorio de esta interleukina aumenta el riesgo de CG (118, 119) y el otro no (120). La causa de esta contradicción no se conoce, aunque es concebible que dada la naturaleza multifactorial del CG, otros factores genéticos de susceptibilidad, el estilo de vida y factores medioambientales puedan atenuar el efecto de este polimorfismo particular.
No obstante la difusión y apoyo de la hipótesis sobre la carcinogénesis gástrica, de múltiples pasos, para algunos es cuestionable que la metaplasia sea una condición premaligna ya que los carcinomas gástricos del tipo difuso como los carcinomas de células en "anillo de sello", aparecen en gastritis no asociadas a metaplasia intestinal ni a atrofia, y por lo tanto, serían más bien alteraciones paraneoplásicas y no preneoplásicas (121). Sin embargo, hace más de una década se demostró que la metaplasia intestinal I y II no tienen riesgo para desarrollar CG, pero el tipo III tiene un riesgo relativo de 4,6 (122). Por otro lado, en un estudio de seguimiento durante 8 a 9 años de 90 pacientes con metaplasia intestinal tipo III, se encontró que solamente uno de ellos desarrolló CG (123) y en otro estudio más reciente, El-Zimaity y col (124) obtuvieron resultados similares al no encontrar carcinomas ni displasia en ninguno de 33 pacientes con metaplasia tipo II y 34 con metaplasia tipo III, durante un seguimiento de 9 años. En Japón, recientemente, Kakinoki y col (125) encontraron que en pacientes infectados con H. pylori, la mayoría de adenocarcinomas gástricos se produjeron existiendo atrofia leve a moderada. Estos últimos estudios mantienen viva la controversia sobre el papel de la metaplasia gástrica como precursora del CG. Con respecto a esto, actualmente hay evidencia de que las células preneoplásicas y neoplásicas provienen de las células progenitoras de la médula ósea) del adulto (CPMO) o "stem cells", que son "atraídas" (homing), hacía la inflamación gástrica crónica, inducida y perpetuada por H. pylori (126-128). Estos nuevos hallazgos no desacreditan el modelo de Correa, sino que por el contrario lo enriquecen y rescatan la importancia del mismo, ya que los diferentes mediadores inflamatorios son fundamentales para la movilización de las CPMO y para la modulación del riesgo de CG (figura 3). La atrofia y la metaplasia son indicadoras de inflamación crónica y permanente, la cual es determinante para llevar al CG, al producir los tres estados de la carcinogénesis como son la iniciación, promoción y progresión (126-128). La inflamación inducida por H. pylori involucra complejas redes moleculares parcialmente conocidas, que generan destrucción tisular constante, culminando en atrofia y metaplasia intestinal, en individuos genéticamente susceptibles y con condiciones medioambientales favorables para este desenlace. La reparación inicial es asumida sin éxito por "stem cell" de sangre periférica que progresivamente es reemplazada por las CPMO que serían la segunda línea de defensa, ante la severa y permanente inflamación y destrucción tisular (82). De igual manera, las CPMO serían el "stem cell" del CG ya que, por su alto grado de plasticidad, pueden diferenciarse en los diversos tipos de células que originarían las diversas estructuras del tumor (129).
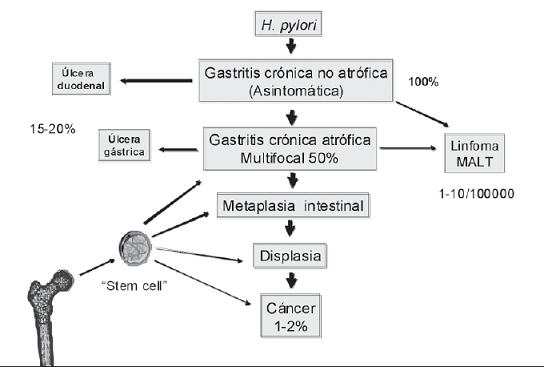
Figura 3. Desenlace de la infección por H. pylori.
Mecanismos directos por H. pylori (factores de virulencia)
H. pylori, genéticamente, es más diverso que la mayoría de las demás poblaciones bacterianas (130). Esta diversidad genética es considerada una característica involucrada en la capacidad del microorganismo para producir diferentes enfermedades. Está más allá del objetivo de trabajo discutir los múltiples factores de virulencia de este microorganismo, por lo cual solo destacaremos algunos de los que consideramos más relevantes.
Isla de patogenicidad y gen CagA
La mayoría de las cepas de H. pylori se pueden agrupar en dos fenotipos distintos con base en la presencia o no de la isla de patogenicidad cag (cag PAI) (108, 110, 130, 131). Esta cag PAI es una región nueva de DNA adquirida por H. pylori en el curso de su evolución mediante transferencia horizontal a partir de otras bacterias (131). Esta isla contiene 30 a 40 genes, incluyendo el gen asociado a la citotoxina (cagA), que codifica la proteína CagA (108, 130, 131). La cag PAI está presente en el 60% de los H. pylori de los países occidentales y en más del 90% de los de países del Asia del este (107, 132). La proteína CagA es altamente inmunogénica, y tiene un peso molecular de 128-140 kDa (130 y las cepas que expresan esta proteína son más virulentas que los que no la expresan (110, 130-132). Las primeras inducen mayor producción de citoquinas inflamatorias como IL-8, y mayor proliferación y apoptosis celular (62, 107, 133). Estas proteínas CagA son "inyectadas por H. pylori, en el interior del citoplasma de la célula epitelial, por un mecanismo secretorio, tipo IV, o "jeringa molecular" (134-136). La translocación de CagA en la célula epitelial gástrica, produce importantes cambios estructurales y funcionales que "benefician" a la bacteria (134). Una vez inyectada, la célula epitelial la reconoce como una molécula de señalización y de manera similar a otras proteínas de señalización, es fosforilada, en grado variable, en unos sitios que contienen la secuencia de cinco aminoácidos Glu-Pro-Ile-Tyr-Ala, denominados motivos EPIYA (130, 134-136). Cuando CagA es fosforilada por estas kinasas, se activa la tirosin fosfatasas SHP-2 una oncoproteína cuya mutación está asociada con procesos malignos en humanos (130-136, 137, 138). CagA desregula a SHP-2 perturbando a la Erk MAP quinasa así como defosforilando FAK kinasas involucradas en adhesión focal induciendo cambios en la morfología celular, la cual adopta un fenotipo conocido como "célula en colibrí" (135-137). CagA también daña la interacción célula-célula de manera independiente a la fosforilación, destruye las uniones estrechas y causa perdida de la polaridad en las células epiteliales y también desestabiliza el complejo E-caderina / β-catenina. (22, 130). Recientemente, se produjo la primera evidencia experimental de que Cag A es una verdadera oncoproteína de H. pylori al demostrar que en ratones transgénicos la fosforilación de CagA, se asoció con la aparición de tumores pero no así cuando se inhibió la capacidad de fosforilarse (139).
A mayor fosforilación de la proteína CagA, mayor potencial oncogénico de la cepa de H. pylori. De acuerdo con los sitios de fosforilación (motivos EPIYA), la proteína CagA, puede ser subclasificada en dos tipos principales (130, 139). CagA del este asiático y CagA occidental (130, 135). El motivo EPIYA, es parte de cuatro distintos sitios EPIYA: EPIYA-A, -B, -C y -D (138, 139). Cada motivo, se define por la secuencia de aminoácidos que rodean la secuencia de EPIYA. En los países occidentales, los CagA más frecuentes son EPIYA-A y B, seguidos por los sitios C repetidos uno a tres veces (ABC, ABCC y ABCCC), de los cuales el más frecuente es el tipo ABC (135). En los países asiáticos, la mayoría de las CagA, son EPIYA-A, y -D (Tipo ABD). La importancia de identificar estos distintos tipos de CagA, reside en que se asocian de manera distinta con la prevalencia y mortalidad de CG. Así, las CagA, de las diversas regiones del mundo con alta prevalencia de CG, es similar al CagA del este asiático y al contrario en donde hay baja prevalencia del tumor, la CagA es tipo occidental (130, 140).
Además de la proteína CagA, el sistema de secreción tipo IV, también libera al interior de las células gástricas pequeñas moléculas efectoras como peptidoglicanos (PGC) de la pared celular (141). En el citoplasma de la célula el PGC es reconocido por la proteína de defensa del sistema inmune innato NOD1, llevando a activación del factor nuclear k beta (NF-kB), el cual aumenta la expresión de genes que codifican proteínas proinflamatorias como la IL-8, quemoquina CXC y defensina B, que es un péptido antimicrobiano (141).
Gen vacA y proteína VacA
La proteína VacA, o citotoxina vacuolizante, es uno de los más importantes factores de virulencia de H. pylori, es codificada por el gen del mismo nombre (vac a), tiene un peso molecular de aproximadamente 139 kDa derivando su nombre de uno de sus primeros efectos estudiados, como es la producción de masiva vacuolización, de las células epiteliales gástricas (110, 142, 143-146). A diferencia del gen cag A, todas las cepas de H. pylori, poseen el gen vac A, pero su expresión funcional es variable, por lo cual, no todas inducen vacuolización de las células epiteliales (110, 142, 143). Si bien este gen está presente en todas las cepas de H. pylori, tiene varios polimorfismos (142, 143). Clásicamente se ha considerado que tiene dos regiones: la región "s" o secuencia de señal y la región media del genoma o "m" y para cada uno de ellas hay dos versiones (alelos): s1 o s2 y m1 o m2, ocurriendo en todas las posibles combinaciones de los dos: s1m1, s1m2, s2m2, etc. Los s1 y m1 a su vez pueden ser subdivididos en s1a, s1b y s1c y el "m" en m1a, m1b y m1c (143-145). Además de la vacuolización celular, la proteína VacA produce otras alteraciones en el epitelio gástrico (110, 147): formación de poros, alteración de las uniones estrechas intercelulares, apoptosis, supresión del sistema inmune del huésped, bloqueo de los fagosomas de los macrófagos, interferencia con la presentación de antígenos a la célula T, inhibición de la activación y proliferación de los linfocitos T ("down regulation"), (110, 146). El genotipo s2 bloquea la actividad vacuolizante en cambio las cepas vac s1 tienen mayor citotoxicidad y se asocian con mayor inflamación gástrica y úlcera duodenal (146). En general las cepas vac s1m1 producen mayores cantidades de citotoxina y están relacionadas con gastritis más severas, con atrofia, metaplasia intestinal, CG y con úlceras gastroduodenales (110, 143-146). En cambio, las cepas con genotipo vac As2m2 son menos virulentas, producen escasa o nula citotoxina, inducen gastritis más leve y se asocian menos a CG (110, 143, 146). Recientemente, Sugimoto describió que en los países latinoamericanos, las cepas con genotipos s1m1 tienen mayor riesgo de CG (145). Para CG el genotipo s1 tiene un OR de 4,17 y el m1 un OR 3,6. Además de estos polimorfismos clásicos del gen vacA, hace poco se describió una nueva región del mismo, denominada región intermedia (i) la cual a su vez tiene dos subregiones: i1 e i2 (147). En estudios previos se había encontrado que, en pacientes con CG, las cepas más frecuentemente encontradas eran las s1m1 y ocasionalmente las cepas s1m2 (145, 146). En este estudio, se encontró que en la región intermedia i1 estaban incluidas todas las cepas s1m1 y todas las cepas s1m2 que tenían actividad vacuolizante in vitro, por lo cual los autores concluyeron que este nuevo genotipo sería el mejor marcador carcinogénico del gen vacA (147).
Otros factores de virulencia
Además de los genes cagA y vacA, H. pylori tiene muchísimos otros factores de virulencia, entre los cuales se encuentran el oipA ("outer membrane inflammatory proteína"), BabA ("Blood Group antigen binding adhesin") y alpAB (60, 107, 110, 148-151). La BabA, es una proteína externa, que le permite a H. pylori adherirse a los antígenos Lewis B, expresados en las membranas de las células epiteliales gástricas (148). Las cepas que expresan estas proteínas producen lesiones más severas. Las respectivas proteínas codificadas por estos genes están estrechamente relacionadas con las patologías gastroduodenales asociadas con H. pylori, incluyendo CG (73, 140).
Hasta el momento no se sabe con exactitud el mecanismo de acción de los múltiples factores de virulencia de H. pylori, y su conocimiento, sus interacciones, así como sus variaciones genéticas podrían contribuir a la identificación de los pacientes con mayor riesgo para CG (59, 73, 80). Por ejemplo, la interacción de las cepas CagA positivas con el polimorfismo 511 de l IL-1B incrementa notablemente el riesgo de CG y la asociación de cagPAI con Oip influye en los niveles de IL-8, ya que ambos se necesitan para activar la región promotora de esta citoquina (150, 151). Otros mecanismos adicionales por los cuales H. pylori promueve la carcinogénesis (152), se muestran en la tabla 2.
Tabla 2. Mecanismos oncogénicos producidos por H. pylori.

Factores genéticos del huésped
Los polimorfismos genéticos del individuo infectado influyen en las diferentes manifestaciones clínicas de la infección por H. pylori. Dentro de estos, previamente mencionamos los polimorfismos de la IL-1B y el IL-1RN los cuales se asocian con mayor riesgo de CG (118-120). Otro factor de riesgo independiente para CG son los genotipos del factor de necrosis tumoral alfa (TNFa) (153). El TNFa es una citoquina, con fuerte actividad proinflamatoria, que también es inducida por H. pylori (8, 60, 93, 96, 108) y al igual que la IL-1B inhibe, pero en menor intensidad, la producción de HCl. El alelo A de esta citoquina tiene un OR para CG de 2,2 (IC 95% 1,4-3,7) (154). La IL-10, por el contrario, es una citoquina antiinflamatoria que produce regulación negativa ("down regulation") de las citoquinas proinflamatorias (IL-1B, TNFa, Interferón G, etc.) y por lo tanto su deficiencia contribuye a aumentar la respuesta inmunológica de tipo Th1 y con ella mayor inflamación gástrica (80, 96, 152, 153). Otros polimorfismos descritos que se asocian con mayor riesgo de CG son los de la región promotora de la IL-8 (155, 156).
Prevención del CG
Teniendo en cuenta la asociación incontrovertible entre H. pylori y CG, la erradicación de la infección podría ser una excelente estrategia para evitar el tumor, pero hasta el momento no se ha demostrado la eficacia de la misma. En la actualidad, hay acuerdo en que el momento óptimo para disminuir el riesgo de CG es eliminar el microorganismo, antes de que exista atrofia y metaplasia intestinal (157-159). Esto fue demostrado por Wong en China (158) y recientemente en dos pacientes a quienes se les erradicó H. pylori cuando ya tenían atrofia y metaplasia intestinal y posteriormente desarrollaron CG (160). Por lo anterior, se considera que la atrofia y metaplasia intestinal son el "punto de no retorno" y la erradicación en ese momento ya no disminuye el riesgo de CG (158-160). Esto implica que la erradicación de la infección no previene el tumor en todos los pacientes y, al existir estas lesiones gástricas avanzadas, es necesario el seguimiento endoscópico para una detección temprana del CG (160). En cambio, en pacientes con linfoma MALT gástrico, la erradicación de H. pylori produce la curación en la mayoría de los pacientes (161).
Un reciente avance trascendental en la prevención de H. pylori es un estudio doble ciego de fase I, de Malfertheiner y col (162), quienes encontraron que una vacuna profiláctica recombinante que utiliza como antígenos la citotoxina VacA, la proteína CagA y la proteína activadora de neutrófilos (NAP), indujo la producción de anticuerpos contra las tres proteínas en el 86% de voluntarios sanos con edades entre 18 y 40 años. La conclusión de los autores es que esta combinación de antígenos tiene una aceptable seguridad e inmunogenicidad, que estimula linfocitos T de memoria y amerita estudios clínicos adicionales.
Conclusiones
En la carcinogénesis por H. pylori, hay dos vías involucradas: mecanismos indirectos a través de la inflamación persistente inducida por la infección, la cual se acompaña de hiperproliferación celular, con alto riesgo y daño del DNA por radicales libres y con participación adicional de células progenitoras de la médula ósea que sería el "stem cell" para el CG. La segunda vía involucra acciones directas de proteínas de H. pylori sobre las células gástricas. A la luz de la información disponible, lo más probable es que ambas vías participan en la génesis del CG (figura 4). Entre los factores genéticos del huésped hay evidencia de que polimorfismos genéticos de IL-1B, TNF, IL-8, INF gama e IL-10 entre otros, inducen una fuerte respuesta inflamatoria que se asocia con mayor riesgo de CG. Finalmente, el CG es una entidad multifactorial, que involucra factores genéticos del individuo, factores medioambientales y la infección por H. pylori, la cual es el factor más importante (figura 5).
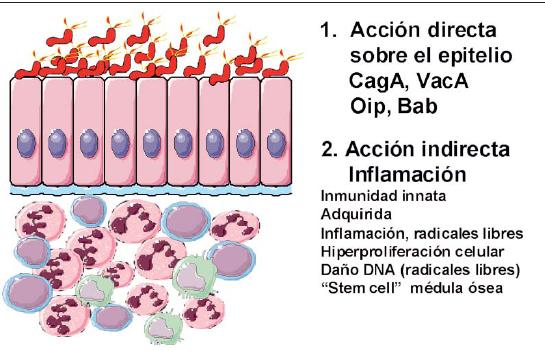
Figura 4. H. pylori y vías de la carcinogénesis.
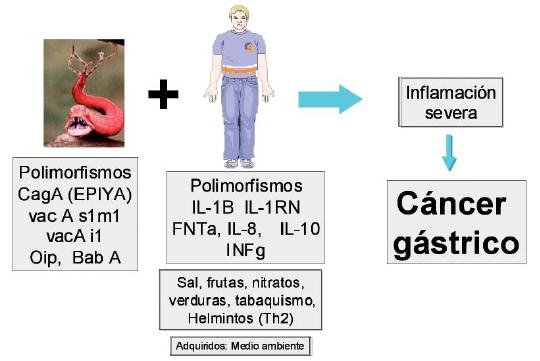
Figura 5. H. pylori y cáncer gástrico (Agente, huésped, medio ambiente).
Referencias
1. Ferlay J, Bray F, Pisani P, et al. GLOBOCAN 2002: Cancer Incidence, Mortality and Prevalence Worldwide, versión 2.0 IARC CancerBase No 5 Lyon: IARC 2004. [ Links ]
2. Parkin DM, Bray F, Ferlay J, Paisani P. Global Cancer Statistics 2005; CA Cancer J Clin 2005; 55: 74-108. [ Links ]
3. Krew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354-62. [ Links ]
4. Hamashima C, Shibuya D, Yamazaki H, Inoue K, Fukao A, Saito Sobue T. The Japanese guidelines for gastric cancer screening. Jpn J Clin Oncol 2008; 38: 259-67. [ Links ]
5. Piñeros M, Hernández G, Bray F. Increasing mortality rates of common malignancies in Colombia. Cancer 2004; 101: 2285-92. [ Links ]
6. Correa P, Piazzuelo MB, Camargo MC. Overview and pathology of gastric cancer. En Wang T, Fox J, Giraud A. (Edit). The biology of gastric cancers. Springer Science Business + Media 2009. p. 21-44. [ Links ]
7. Smith MG, Hold GL, Tahara E, El-Omar EM. Cellular and molecular aspects of gastric cancer. World J Gastroenterol 2006; 12: 2979-90. [ Links ]
8. Mosss SF, Malfertheiner P. Helicobacter and gastric malignancies. Helicobacter 2007; 12(Suppl 1): 23-30. [ Links ]
9. Farinha P, Gascoyne RD. Helicobacter pylori and MALT lymphoma. Gastroenterology 2005; 128: 1579-1605. [ Links ]
10. Jarvi O, Lauren P. On the role of heterotopias of the intestinal epithelium in the pathogenesis of gastric cancer. Acta Pathol Microbiol Scand 1951; 29: 26-44. [ Links ]
11. Lauren, P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31-49. [ Links ]
12. Correa P, Carneiro F. Classification of gastric carcinomas. Curr Diagn Pathol 1997; 4: 51-9. [ Links ]
13. Ekstrom AM, Serafini M, Nyren O, Hansson LE, Ye W, Wolk A. Dietary antioxidante intake and the risk of noncardia cancer for the intestinal and diffuse types: a population-based case-control study in Sweden. Int J Cancer 2000; 87: 133-40. [ Links ]
14. El-Omar EM, Lochhead P. Gastric cancer. Br Med Bull 2008; 85: 87-100. [ Links ]
15. Henson DE, Dittus C, Younes M, Nagyen H, Albores-Saavedra J. Differential trends in the intestinal and diffuse topless of gastric carcinoma in the United States-2000: increase in the signet ring cell type. Arch Pathol Lab Med 2004; 128: 765-70. [ Links ]
16. Oliveira C, Seruca R, Carneiro F. Hereditary gastric cancer. Best Pract Res Clin Gastroenterol 2009; 23: 147-57. [ Links ]
17. Alberts SR, Cervantes A, van de Velde CJ. Gastric cancer: epidemiology, pathology and treatment. Ann Oncol 2003; 14 (suppl 2): ii31-ii36. [ Links ]
18. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392: 402-5. [ Links ]
19. Carneiro F, Sobrinho-Simoes M. Hereditary diffuse gastric cancer: lessons from histopatholgy. Adv Anat Pathol 2005; 12: 151-2. [ Links ]
20. Medina-Franco H, Barreto-Zúñiga R, García-Álvarez MN. Preemptive total gastrectomy for hereditary gastric cancer. J Gastrointest Surg 2007; 11: 314-7. [ Links ]
21. Medina-Franco H, Medina AR, Vizcaíno G, Medina-Franco JL. Single nucleotide polymorphisms in the promoter region of the E-cadherin gene in gastric cancer: case-control study in a young Mexican population. Ann Surg Oncol 2007; 14: 2246-9. [ Links ]
22. Cisco RM, Ford JM, Norton JA. Hereditary diffuse gastric cancer. Cancer 2008; 113 (7 Suppl): 1850-6. [ Links ]
23. Masciari S, Larsson N, Senz J, Boid N, Kaurah P, Kandel MJ, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet 2007; 44: 726-31. [ Links ]
24. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19 (Suppl.1): S37-S43. [ Links ]
25. Kabir S. Effect of Helicobacter pylori eradication on incident of gastric cancer in human and animal models: underlying biochemical and molecular events. Helicobacter 2009; 14: 159-71. [ Links ]
26. Correa P, Chen VW. Gastric Cancer. Cancer Surv 1994; 19: 55-76. [ Links ]
27. Kono S, Irohata T. Nutrition and stomach cancer. Cancer Causes Control 1996; 7: 41-55. [ Links ]
28. Larsson SC, Bergkvist L, Wolk A. Fruit and vegetable consumption and incidence of gastric cancer: A prospective study. Cancer Epidemiol Biomarkers Prev 2006; 15: 1998-2001. [ Links ]
29. Bae JM, Lee EJ, Guyatt G. Citrus fruit intake and stomach cancer risk: a quantitative systematic review. Gastric Cancer 2008; 11: 23-37. [ Links ]
30. González CA, Pera G, Agudo A, Bueno-de Mezquita HB, Ceroti M, et al. Fruit and vegetable intake and the risk of stomach and oesophagus adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC-Eurogast). Int J Cancer 2006; 118: 2559-66. [ Links ]
31. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for preventing gastrointestinal cancers. Cochrane Database of Systematic Reviews 2004; Issue 4. [ Links ]
32. Liu C, Huang XD, Russell RM. Diet and gastric cancer. En Wang TC, Fox J, Giraud A. (Edits). The biology of gastric cancers. Springer Science+Business Media LLC 2009. p. 59-89. [ Links ]
33. Correa P, Fontham ET, Bravo JC, Bravo LE, Ruiz B, Zarama G, Realpe JL, Malcom GT, Li D, Johnson WD, Mera R. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-helicobacter pylori therapy. J Natl Cancer Inst 2000; 92: 1881-8. [ Links ]
34. Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005; 54: 1536-40. [ Links ]
35. Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ, Yang CS, Zheng SF, Gail M, Li GY, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst 1993; 85: 1483-92. [ Links ]
36. Goswami UC, Sharma N. Efficiency of a few retinoids and carotenoids in vivo in controlling benzo[a]pyrene-induced forestomach tumour in female Swiss mice. Br J Nutr 2005; 94: 540-3. [ Links ]
37. Velmurugan B, Bhuvaneswari V, Burra UK, Nagini S. Prevention of N-methyl-N′-nitro-N-nitrosoguanidine and saturated sodium chloride-induced gastric carcinogenesis in Wistar rats by lycopene. Eur J Cancer Prev 2002; 11: 19-26. [ Links ]
38. Velmurugan B, Bhuvaneswari V, Nagini S. Antiperoxidative effects of lycopene during N-methyl-N′-nitro-N-nitrosoguanidine-induced gastric carcinogenesis. Fitoterapia 2002; 73: 604-11. [ Links ]
39. Velmurugan B, Mani A, Nagini S. Combination of S-allylcysteine and lycopene induces apoptosis by modulating Bcl-2, Bax, Bim and caspases during experimental gastric carcinogenesis. Eur J Cancer Prev 2005; 14: 387-93. [ Links ]
40. Liu C, Russell RM, Wang XD. Lycopene supplementation prevents smoke-induced changes in p53, p53 phosphorylation, cell proliferation, and apoptosis in the gastric mucosa of ferrets. J Nutr 2006; 136: 106-11. [ Links ]
41. Wang X, Willen R, Wadstrom T. Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob Agents Chemother 2000; 44: 2452-7. [ Links ]
42. Liu BH, Lee YK. Effect of total secondary carotenoids extracts from Chlorococcum sp on Helicobacter pylori-infected BALB/c mice. Int Immunopharmacol 2003; 3: 979-86. [ Links ]
43. World Cancer Research Fund, American Institute for Cancer Research. Food, nutrition and the prevention of cancer: a global perspective. Washington, DC: American Institute for cancer Research 1997. [ Links ]
44. Kato S, Tsukamoto T, Mizoshita T, Tanaka H, Kumagai T, Ota H, Katsuyama T, Asaka M, Tatematsu M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer 2006; 119: 1558-66. [ Links ]
45. Watanabe H, Takahashi T, Okamoto T, Ogundigie PO, Ito A. Effects of sodium chloride and ethanol on stomach tumorigenesis in ACI rats treated with N-methyl-N′-nitro-Nnitrosoguanidine: a quantitative morphometric approach. Jpn J Cancer Res 1992; 83: 588-93. [ Links ]
46. Furihata C, Sato Y, Hosaka M, Matsushima T, Furukawa F, Takahashi M. NaCl induced ornithine decarboxylase and DNA synthesis in rat stomach mucosa. Biochem Biophys Res Commun 1984; 121: 1027-32. [ Links ]
47. Sorbye H, Kvinnsland S, Svanes K. Effect of salt-induced mucosal damage and healing on penetration of N-methyl-N′-nitro-N-nitrosoguanidine to proliferative cells in the gastric mucosa of rats. Carcinogenesis 1994; 15: 673-9. [ Links ]
48. Fox JG, Dangler CA, Taylor NS, King A, Koh TJ, Wang TC. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res 1999; 59: 4823-8. [ Links ]
49. Jakszyn P, González, CA. Nitrosamine and related food intake and gastric and oesophageal cancer risk: a systematic review of the epidemiological evidence. World J Gastroenterol 2006; 12: 4296-303. [ Links ]
50. Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett.1995; 93: 17-48. [ Links ]
51. Jakszyn P, Bingham S, Pera G, Agudo A, Luben R, Welch A, et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis.2006; 27: 1497-501. [ Links ]
52. Schistosomes, Liver fluyes and Helicobacter pylori IARC Working group on the evaluation of carcinogenic risks to humans. Lyon, 7-14 June 1994. IARC Monog Eval Carcinog Risks Hum 1994; 61: 1-41. [ Links ]
53. Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, et al. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 1991, 302: 1302-5. [ Links ]
54. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentrich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127-31. [ Links ]
55. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser M, et al. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132-6. [ Links ]
56. Sugiyama A, Maruta F, Ikeno T, Ishida K, Kawasaki S, Katsuyama T, et al. Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 1998; 58: 2067-69. [ Links ]
57. Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998; 115: 642-8. [ Links ]
58. An International association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993; 341: 1359-62. [ Links ]
59. Huang JQ, Zheng GF, Sumanak K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cag A seropositivity and gastric cancer. Gastroenterology 2003; 125: 1636-44. [ Links ]
60. Peter S, Begglinnger C. Helicobacter pylori and gastric cancer: The causal relationship. Digestion 2007; 75: 25-35. [ Links ]
61. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med 2002; 347: 1175-86. [ Links ]
62. Mueller A, Falkow S, Amieva MR. Helicobacter pylori and gastric cancer: what can be learned by studying the response of gastric epithelial cells to the infection? Cancer Epidemiol Biomarkers Prev 2005; 14: 1859-64. [ Links ]
63. Peter S, Beglinger C. Helicobacter pylori and gastric cancer: the causal relationship. Digestion 2007; 75: 25-35. [ Links ]
64. Wu MS, Shun CT, Wu CC, et al. Epstein Barr virus associated with Helicobacter pylori. Infection and genetic alterations. Gastroenterology 2000; 118: 1031-8. [ Links ]
65. Report of the Digestive Health Initiative International Update. Conference on Helicobacter pylori. Gastroenterology 1997; 113 (Suppl): S4-S8. [ Links ]
66. Saad R, Chey W. A clinicians guide to managing Helicobacter pylori infection. Clev Clin J Med 2005; 72: 109-124. [ Links ]
67. Parsonnet J. Helicobacter pylori: the size of the problem. Gut 1998; 43: S6-S9. [ Links ]
68. Correa P, Schneider BG. Etiology of gastric cancer: what is new? Cancer Epidemiol Biomarkers Prev 2005; 14: 1865-8. [ Links ]
69. Guillen D, McColl KEL. Gastroduodenal disease, Helicobacter pylori, and genetic polymorphisms. Clinical Gastroenterol Hepatol 2005; 3: 1180-86. [ Links ]
70. El Omar EM, Penman I, Ardill JE, Chittajallu RS, Howie C, Mc Coll KE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995; 109:681-91. [ Links ]
71. Lai LH, Sung JJY. Helicobacter pylori and benign upper digestive disease. Best Pract Res Clin Gastroenterol 2007; 21: 261-79. [ Links ]
72. Faber K. Chronic gastritis: its relation to achylia and ulcer. Lancet 1927; 2: 902-7. [ Links ]
73. Graham DY, Lu H, Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: facts or medical myths. J Dig Dis 2009; 10: 77-84. [ Links ]
74. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784-9. [ Links ]
75. Chiba T, Seno H, Marusawa Y, Okazaki K. Host factors are important in determining clinical outcomes of Helicobacter pylori infection. J Gastroenterol 2006; 41: 1-9. [ Links ]
76. Chiba T, Marusawa H, Seno H, Watanabe N. Mechanism Fort he gastric cancer development by Helicobacter infection. J Gastroenterol Hepatol 2008; 23: 1175-81. [ Links ]
77. Bonnevie O. The incidence of duodenal ulcer in Copenhagen County. Gastroenterology 1975; 10: 385-93. [ Links ]
78. Hu PJ, Li YY, Zhou MH, Chen MH, Huang BJ, Mitchel HM, et al. Helicobacter pylori associated with a high prevalence of duodenal ulcer disease and low prevalence of gastric cancer in a developing nation. Gut 1995; 36: 198-202. [ Links ]
79. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg pathol 1995; 19: S37-S43. [ Links ]
80. Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori Basic research to patient care. Gastroenterology 2006; 130: 188-206. [ Links ]
81. Matsukura N, Suzuki K, Kavachi T, Aoyagi M, Sugimura T, Kitaoka H, et al. Distribution of marker enzymes and mucin in intestinal metaplasia in human stomach and relation to complete and incomplete types of intestinal metaplasia to minute gastric carcinomas. J Natl Cancer Inst 1980; 65: 231-40. [ Links ]
82. Correa P, Hoghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007; 133: 659-72. [ Links ]
83. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 195-200. [ Links ]
84. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T. Helicobacter pylori infection triggers aberrant expresión of activation-induced cytidine deaminase in gastric epithelium. Nat Med 2007; 13: 470-6. [ Links ]
85. Takaishi S, Wang TC. Providing AID to p53 mutagenesis. Nat Med 2007; 13: 404-6. [ Links ]
86. Correa P, Haenszel W, Cuello C, Tannembaum S, Archer M. A model for gastric cancer epidemiology. Lancet 1975; 2: 58-60. [ Links ]
87. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 1273-5. [ Links ]
88. Correa P. The epidemiology and pathogenesis of chronic: three etiologic entities. Front Gastrointest Res 1980; 6: 98-108. [ Links ]
89. Correa P. Clinical implications of recent development in gastric cancer pathology and epidemiology. Sem Oncol 1985; 12: 2-10. [ Links ]
90. Bonne C, Hartz PH, Klerks JV, Posthuma JH, Radsma W, Tjokronegoro S, et al. Morphology of the stomach and gastric secretion in Malays and Chinese and the different incident of gastric ulcer and cancer in these races. Am J Cancer 1938; 33: 265-79. [ Links ]
91. Siurala M, Varis K, Wiljasalo M. Studies of patients with atrophic gastritis: a 10 -15 year follow-up. Scand J Gastroenterol 1966; 1: 40-8. [ Links ]
92. Volk J, Parsonnet J. Epidemiology of Gastric Cancer. En Wang TC, Fox J, Giraud A. (Eds). The biology of Gastric cancers. Springer Science+ Business Media LLC 2009. p. 25-57. [ Links ]
93. Matysiak-Budnik T, Mégraud F. Helicobacter pylori infection and gastric cancer. Eur J Cancer 2006; 42: 708-16. [ Links ]
94. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. NodI detect a unique muropeptide from gram negative bacterial peptidoglycan. Science 2003; 300: 1584-87. [ Links ]
95. Girardin SE Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, et al. Peptiglycan molecular requeriments allowing detection by NodI and Nod2. J Biol Chem 2003; 278: 41702-8. [ Links ]
96. Keeffe J, Moran AP. Conventional, regulatory and unconventional T cells in the immunologic response to Helicobacter pylori. Helicobacter 2008; 13: 1-19. [ Links ]
97. Ma J, Chen T, Mandelin J, Ceponis A, Miller NE. Regulation of macrophage activation. Cell Mol Life Sci 2003; 60: 2334-46. [ Links ]
98. Malaty HM. Epidemiology of Helicobacter pylori. Best Pract Res Clin Gastroenterol 2007; 21: 205-14. [ Links ]
99. Hold G, Rabkin CS, Chow WH, Smith MG, Gammon MD, Sisch HA, et al. A gunctional polymorphisms of Toll like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 2007; 132: 905-12. [ Links ]
100. Hisida A, Matsuo K, Goto Y, Mitsuda Y, Hiraki K, Naito M, et al. Toll-like Receptor 4+3725G/C polymorphisms, Helicobacter pylori seropositivity and the risk of gastric atrophy and gastric cancer. Helicobacter 2009; 14: 47-53. [ Links ]
101. D´Elios MM, Amedi A, Benagiano M, Azzurri A, Del Prete G. Helicobacter pylori, T cells and cytokines: the "dangerous liaisons". FEMS Immunol Med Microbiol 2005; 44: 113-9. [ Links ]
102. Lehmann FS, Terracciano L, Carena I, Baeriswyl C, Drewe J, Tornillo L, et al. In situ correlation of cytokine secretion and apoptosis in Helicobacter pylori-associated gastritis. Am J Physiol Gastrointest Liver Physiol 2002; 283: G481-G488. [ Links ]
103. Holcombe C. Helicobacter pylori: The African enigma. Gut 1992; 33: 429-31. [ Links ]
104. Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, et al. The prevalence of Th17 cells in patients with gastric cáncer. Bioch Bioph Res Com 2008; 374: 533-7. [ Links ]
105. Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of Cag A phosphorilation for clinical aislates of Helicobacter pylori. Gastroenterology 2004; 127: 514-23. [ Links ]
106. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo C, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 95-200. [ Links ]
107. Wu MS, Chen CJ, Lin JT. Host environment interactions: their impact on progression from gastric inflammation to carcinogenesis and the development to new approaches to prevent and treat gastric cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 1878-82. [ Links ]
108. Amieva MR, El-Omar EM. Host bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008; 134: 306-23. [ Links ]
109. Ferreira AC, Isomoto H, Moriyama M, Fujioka T, Machado JC, Yamaoka Y. Helicobacter and gastric malignancies. Helicobacter 2008; 13 (Suppl 1): 28-34. [ Links ]
110. Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321-33. [ Links ]
111. Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol 1991; 261: G559-64. [ Links ]
112. El-Omar EM, Carrington M, Chow WH, McColl KEL, Bream JH, Young HA, et al. Interleukin-1 plymorphims associated with increased risk of gastric cancer. Nature 2000; 404: 398-402. [ Links ]
113. Machado JC, Pharoah P, Sousa S, Carvalho R, Oliveira C, Figuereido C, et al. Interleukin-1b and interleukin-1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001; 121: 823-9. [ Links ]
114. Chen A, Li CN, Hsu PI, Lai KH, Tseng HH, Hsu PN, et al. Risks of interleukin-1 genetic polymorphisms and Helicobacter pylori infection in the development of gastric cancer. Aliment Pharmacol Ther 2004; 20: 203-11. [ Links ]
115. Lahner E, Corleto VD, D´Ambra G, Di Giulio E, D´Elle Fave G, Anibale B. Is Interleukin 1 genotyping useful for the clinical management of patients with atrophic body gastritis? Aliment Phrmacol ther 2008; 27: 355-65. [ Links ]
116. Palli D, Saieva C, Luzzi I, Masala G, Topa S, Sera F, et al. Interleukin- 1 gene polymorphisms and gastric cancer risk in a high-risk Italian population. Am J Gastroenterol 2005; 100: 1941-8. [ Links ]
117. Lee SG, Kim B, Choi W, Lee I, Choi J, Song K. Lack of association between pro-inflammatory genotypes of the interleukin-1 (IL-1B -31 C/+ and IL-1RN *2/*2) and gastric cancer/duodenal ulcer in Korean population. Cytokine 2003; 21: 167-71. [ Links ]
118. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: a meta-analysis. Int J cancer 2007; 120: 552-62. [ Links ]
119. Camargo MC, Mera R, Correa P, Peek Jr RM, Fontham ET, Goodman KJ, et al. Interlukin-1beta and antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1674-87. [ Links ]
120. Kamangar F, Cheng C, Abnet CC, Rabkin C. Interleukin -1B polymorphisms and gastric cancer risk-A Metanalysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1920-8. [ Links ]
121. Meining A, Morgner A, Miehlke S, Morgner A, Bayerdorffer E, Stolte M. Atrophy-metaplasia-dysplasia-carcinoma sequence in the stomach: a reality or merely a hypothesis? Best Pract Res Gastroenterol 2001; 15: 983-98. [ Links ]
122. Filipe MI, Muñoz N, Matko I, Kato I, Pompe-Kim V, Jutersek A, et al. Intestinal metaplasia types and the risk of gastric cancer: A cohort in Slovenia. Int J Cancer 1994; 57: 324-9. [ Links ]
123. Ectors N, Dixon MF. The pronostic value of sulphomucin positive intestinal metaplasia in the deveopment of gastric cancer. Histopahology 1986; 10: 1271-7. [ Links ]
124. El Zimaity HM, Ramchatesingh J, Saeed MA, Hraham DY. Gastric intestinal metaplasia: subtypes and natural history. J Clin Pathol 2001; 54: 679-83. [ Links ]
125. Kakinoki R, Kushima R, Matsubara A, Saito Y, Okabe H, Fujiyama Y et al. Re-evaluation of histogenesis of gastric carcinoma: A comparative histopathological study between Helicobacter pylori-negative and H. pylori-positive cases. Dig Dis Sci 2009; 54: 614-20. [ Links ]
126. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrowderived cells. Science 2004; 306: 1568-71. [ Links ]
127. Li HC, Stoicov C, Rogers AB, Houghton J. Stem cells and cancer: evidence for bone marrow stem cells in epithelial cancers. World J Gastroenterol 2006; 12: 363-71. [ Links ]
128. Houghton J, Morozov A, Smirnova I, Wang TC. Stem Cell and Cancer. Seminars Cancer Biol 2007; 17: 191-203. [ Links ]
129. Takaishi S, Okumura T, Wang TC. Gastric cancer Stem Cell. J Clin Oncol 2008; 26: 2876-82. [ Links ]
130. Azuma T. Role of Cag A in the Helicobacter pylori infection and pathology. En Wang TC, (Eds). The biology of gastric cancers. Springer Science+Business Media, LLC 2009. p. 389-401. [ Links ]
131. Harcker J, Kaper JB. Pathogenicity Islands and the Evolution of Microbes. Ann Rev Microbiol 2000; 54: 641-679. [ Links ]
132. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. Cag A pathogenicity islad of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U.S.A 1996; 93: 14648-53. [ Links ]
133. Crowe SE. Helicobacter pylori infection, chronic inflammation and the development of malignancy. Curr Op Gastroenterol 2005; 21: 32-38. [ Links ]
134. Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 1573-81. [ Links ]
135. Handa O, Naito Y, Yoshikawa T. CagA protein of Helicobacter pylori: A hijacker of gastric epithelial cell signalin. Bioch Pharmacol 2007; 73: 1697-1702. [ Links ]
136. Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA Phosphorylation-Independent function in epithelial proliferation and inflammation. Cell Host Micr 2009; 5: 23-34. [ Links ]
137. Steffen Backert and Matthias Selbach. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 8: 1573-1581. [ Links ]
138. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4: 688-94. [ Links ]
139. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natnl Acad Sci 2008; 105: 1003-8. [ Links ]
140. Yamaoka Y, Kato M, Asaka M. Geographic differences in gastric cancer can be explained by differences between Helicobacter pylori Straits. Intern Med 2008; 47: 1077-83. [ Links ]
141. Fox JG, Wang TC. Inflammation atrophy and gastric cancer. J Clin Invest 2007; 17: 60-9. [ Links ]
142. Cover TL, Tummuru MKR, Cao P, Thompson SA, Blaser MJ. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori Straits. J Biol Chem 1994; 269: 1566-573. [ Links ]
143. Yuan JP, Li T, Chen HB, Li ZH, Yang GZ, Hu BH, et al. Analysis of gene expression profile in gastric cancer cells stimulated with Helicobacter pylori isogenic strains. J Med Microbiol 2004; 53: 965-74. [ Links ]
144. Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori peptic ulceration. J Biol Chem 1995; 270: 1771-7. [ Links ]
145. Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infet 2009 Early Rel (acceso junio 10, 2009). [ Links ]
146. Letley DP, Atherton JC. Natural Diversity in the n terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J Bacteriol 2000; 182: 3278-80. [ Links ]
147. Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh M, et al. A new Helicobacter pylori Vacuolating cytotoxin determinant the intermediate region is associated with gastric cancer. Gastroenterology 2007; 133: 926-36. [ Links ]
148. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesion binding fucosylated histo-blood group antigens revealed by retagging. Science 1998; 279: 373-7. [ Links ]
149. Peek Jr RM, Thompson SA, Donahue JP, Tham KT, Athertton JC, Blaser MJ, et al. Adherence to gastric epithelial cells induces expression of a Helicobacter pylori gene ice A, that is associated with clinical outcome. Proc Assoc Am Phys 1998; 110: 531-44. [ Links ]
150. Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsiveelement-like element in inteleukin8 promoter in Helicobacter pylori infection. Gastroenterology 2004; 126: 1030-45. [ Links ]
151. Yamaoka Y, Kwon DH, Graham DY, A M (r). 34 000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA 2000; 97: 7533-8. [ Links ]
152. Kontouras J, Zavos C, Chatzopoulos D, Katsinelos P. New aspects of Helicobacter pylori infection involvement in gastric oncogenesis. J Surg Res 2008; 146: 149-58. [ Links ]
153. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003; 124: 1193-1201. [ Links ]
154. Beales IL, Calam J. Interleukin 1 beta and tumor necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998; 42: 227-34. [ Links ]
155. Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor (alpha) and risk of gastric cancer in a Chinese population. Carcinogenesis 2005; 26: 631-6. [ Links ]
156. Ohyauchi M, Imatani A, Yonechi M, Asano N, Miura A, Lijima K, et al. The polymorphisms inerleukin 8-251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut 2005; 54: 330-5. [ Links ]
157. Malfertheiner P, Megraud F, O´Morain C Bazzoli F, El-Omar E, Graham DY, et al. Current concepts in the management of Helicobacter pylori infection- The Maastricht III consensus report. Gut 2007; 56: 772-81. [ Links ]
158. Wong BC, Lam FK, Wong WM, Chen JS, Zheng TT, Fen RE, et al. Helicobacter pylori eradication to prevent gastric cancer in high risk region in China: a randomized controlled trial. JAMA 2004; 291: 187-94. [ Links ]
159. Lochhead P, El-Omar EM. Helicobacter pylori infection and gastric cancer. Best Pract Res Clin Gastroenterol 2007; 21: 281-97. [ Links ]
160. De Vries AC, Kuipers EJ, Raws EAJ. Helicobacter pylori eradication and gastric cancer: when is the horse out of the barn? Am J Gastroenterol 2009; 104: 1342-5. [ Links ]
161. Zullo A, Hassan C, Andriani A, Cristoffari F, De Francesco V, Ierardi E, et al. Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: A pooled Data Analysis. Am J Gastroenterol 2009 advance online publication 16 june 2009. [ Links ]
162. Malferthteiner P, Schulktze V, Rosenkranz B, Kauffman SHE, Ulrichz T, Novicki D, et al. Safety and inmunogenecity of an intramuscular Helicobacter pylori vaccine in noninfected volunteers: a phase I study. Gastroenterology 2008; 135: 787-95. [ Links ]

