










Servicios Personalizados
Revista
Articulo
 texto en
texto en  Inglés (pdf)
Inglés (pdf)
 Articulo en XML
Articulo en XML Referencias del artículo
Referencias del artículo
 Enviar articulo por email
Enviar articulo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Accesos
Accesos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares en
SciELO
Similares en
SciELO -
 Similares en Google
Similares en Google
Compartir

 Permalink
PermalinkRevista colombiana de Gastroenterología
versión impresa ISSN 0120-9957versión On-line ISSN 2500-7440
Rev Col Gastroenterol v.24 n.3 Bogotá jul./sep. 2009
Gastric carcinogenesis
William Otero Regino, MD(1), Martín A. Gómez MD(2), Denny Castro MD(3)
(1) Associate Professor of Medicine, Gastroenterology unit, Universidad Nacional de Colombia, Gastroenterologist at the Clínica Fundadores de Fundación Hospital San Carlos and at the Clínica Carlos Lleras Restrepo, Bogotá, Colombia.
(2) Professor of Medicine, Gastroenterology Unit, Universidad Nacional de Colombia, Hospital El Tunal. Gastroenterologist at the Hospital El Tunal, Fundacion Hospital San Carlos y Clínica Carlos Lleras Restrepo, Bogotá, Colombia.
(3) General Director and Director of Graduate Studies of Gastroenterology at the Centro de Control de Cáncer Gastrointestinal "Dr Luis E Anderson" San Cristobal, Estado Táchira Venezuela.
Received: 02-02-09 Accepted: 12-08-09
Summary
Gastric cancer is second among cancers as a cause of death. More than 90% of all gastric cancers are adenocarcinomas whose principal cause is Helicobacter pylori. Although H. Pylori is a necessary condition, it is not a sufficient condition since only 1-2% of those infected develop gastric cancer. There are multiple factors besides H. Pylori infection involved in the etiology of this cancer. They include genetic factors related to the individual and environmental factors. Although the ways in which H. Pylori participates in this carcinogenesis are not completely clear, two different mechanisms are involved. H. Pylori infection induces persistent inflammation accompanied by hyperproliferation of cells, and it causes damage to DNA from free radicals in which progenitor cells from the bone marrow participate. These cells could be the "stem cells" of gastric cancer. The second path involves the direct action of proteins from H. Pylori on gastric cells. Among the genetic factors involved there is evidence that IL-1B, TNF, IL-8, and INF gama e IL-10 polymorphisms, among others, induce B inflammatory responses which are associated with higher risks of gastric cancer.
Key words
Helicobacter pylori, carcinogenesis, stem cell, polymorphisms
Gastric cancer (GC) is heterogeneous and highly prevalent globally. In 2002 it was estimated that there were 900,000 new cases and 700,000 deaths (1). Overall, it is the fourth most common cancer and second leading cause of cancer deaths (2), explaining 10% of them (3). In Japan (4) and Colombia (5) it is the leading cause of cancer death. In Colombia its incidence is approximately 10 times higher than in the USA (5). Over 90% of GCs are adenocarcinomas (6.7). The rest are less frequently occurring tumors such as lymphomas, gastrointestinal stromal tumors (GIST) and carcinoid tumors (7). The primary etiologic agent of distal or non-cardial GCs and MALT lymphoma is Helicobacter pylori (H. pylori) (8). Gastric cancer develops in 1 to 2% of the people infected with these bacteria, while low grade malignancy MALT lymphoma occurs in 0.7 to 0.8 per 100,000 individuals. This is in contrast to the USA, where 1 case per 30,000 to 80,000 individuals occurs, and to Italy where up to 13 cases occur per 100,000 individuals (9).
According to the classification of Finnish pathologists Jarvi and Lauren, GCs are divided into two histological types: intestinal and diffuse (10.11). They have clear differences in terms of epidemiological, histopathological, endoscopic, clinical and pathogenetic points of view (12). Most CGs are sporadic, but approximately 10% are grouped in families and 10 to 30% of these are inherited (13). The intestinal type is most frequent in sites with high GC prevalence, have better prognoses and occur more often in men over 50 years of age. However, in places with high prevalence it appears at an earlier age (6.7). In places with low prevalence or low risk, as in many developed countries, its incidence has declined in recent decades (7). This GC type appears in stomachs that have atrophic gastritis and intestinal metaplasia. There are higher numbers of cases in underdeveloped countries (6, 7, 14).
In contrast the diffuse type shows no geographic variation, is more common in women, often appears in young people with positive family history, has a worse prognosis and is not associated with gastric atrophy or intestinal metaplasia (6, 14). Its incidence has not changed and even seems to be increasing (15, 16). Esophagogastroduodenoscopy, the best method for GC diagnosis, often has difficulties in the diagnosis of diffuse GC because this cancer tends to follow a submucosal pattern rather than forming exophytic masses (16, 17). The diagnosis is even more difficult when there is linitis plastica.
1-3% of all GCs are inherited. This subset represents a clearly defined clinicopathologic syndrome, known as hereditary diffuse GC (17, 18). In 30% of the families with this entity, mutations have been found in the epithelial cadherin gene (E-cadherin or CDH1) which is an encoded cell adhesion protein (19-22). The mutation is autosomal dominant with 70% penetrance, to be precise the patient has a 70% risk of suffering from GC. Women with this mutation have increased risks of lobular breast cancer (19, 22). This GC has high mortality at an early age. When diagnosed, most patients already have advanced disease and high risks of post surgical relapse (16). In individuals with CDH1 mutations, prophylactic gastrectomy is recommended. For those who do not accept this procedure, upper gastrointestinal endoscopy every 6-12 months is recommended (22). Today, it is recommended that patients be checked for CDH1 mutations in the following situations (22):
Families with two or more cases of diffuse GC, one of them, diagnosed before the age of 50.
Families with more than three cases of diffuse GC, diagnosed at any age.
Isolated individuals with a diagnosis of diffuse GC before 35 years of age.
Isolated individuals with lobular breast cancer and diffuse GC.
Families with a member with diffuse GC and another with breast cancer or colon cancer with signet-cell ring. It is also recommended that families with multiple cases of lobular breast cancer with or without cases of diffuse CG be tested, since CDH1 mutations have also been detected in families with only lobular breast cancer (23).
Taking into account that most GC cases are intestinal, this article will focus on the carcinogenic pathways involved in these cases and will not deal with other tumors. The inspiration for this review came from Doctors Pelayo Correa and David Graham, two universally known figures who have contributed substantially to the understanding of this important disease.
Gastric cancer is a multifactorial disease that develops through a multi-step process that can last 20 years or more (24). Its genesis is complex and involves the participation of three main factors: environmental factors, the agent (H. pylori) and host genetic factors (6, 7.12, 14, 24, 25).
Environmental factors
Diet: Multiple observational studies have examined the association between fruit and vegetable consumption and risk of GC. Some have found that fresh fruit, vitamin C and beta carotene are associated with reduced risk of GC (26-29) although others have not found this effect (30). A Cochrane review concluded that there is no evidence that dietary supplementation with antioxidants, including vitamin C, reduces the risk of GC (31). However, no clinical trials have studied the effectiveness of vegetable and fruit consumption on GC risk. The evidence for its protective effect comes only from epidemiological studies (32).
The results of clinical trials with betacarotene and other antioxidants for prevention of GC are not consistent. In Colombia, Correa et al (33) found that a supplement of 6 mg of beta carotene daily for six years significantly increased the regression rate of gastric atrophy and intestinal metaplasia. However, long-term monitoring of patients in this trial found that the benefit of vitamin C and beta carotene disappeared with time, unlike what had happened in the eradication of H. pylori (34). These results correlate with the work of Blot et al in China which found that supplementation with beta carotene (15 mg), alpha-tocopherol (30 mg) and selenium 50 ugr for 5 years did not alter the mortality rates of patients with cancer of the gastric cardia nor of distal or non-cardia gastric cancer (35).
Experimentally, carotenoids (lycopene, lutein and B-carotene) and retinoids inhibit the growth of chemically induced gastric tumors in laboratory animals (36-39). It is believed that the benefits of carotenoids in GC genesis result from mechanisms such as decreased cell proliferation while simultaneously an antioxidant effect blocks free radicals, preventing oxidative DNA damage (37, 39) and induced apoptosis (40), decreases the H. pylori bacteria population and reduces inflammation by correcting the immune response to Th2 (rather than the wrong' answer that paradoxically is Th1) (41, 42).
Salt: There is evidence of increased GC risk in individuals who have a high salt intake or a high intake of foods preserved in salt (7.9). In an investigation by the World Cancer Research Foundation (WCRF) and the American Institute for Cancer Research (AICR) (43), eight studies found increased GC risk (OR 2.1 to 5.0) with salt consumption. However, four studies found no association. Experimentally salt increases gastric tumors (44, 45). High salt concentrations in the stomach produce various harmful effects on the stomach including inflammation and damage to the mucus layer.
Nitrate, nitrites and nitrosamines
Humans are exposed to two sources of nitrosamines. The first source is preformed nitrosamines present in meats, fish and other foods preserved with nitrites such as pickled, smoked, and salted food and in drinks like beer and whiskey (49). The second source is the vegetable nitrates used as additives in cured meats and cheeses (50). Nitrates in the diet can be reduced to nitrites by bacteria in the oral cavity and these reduced to N-nitroso compounds by bacteria in the stomach in reactions with amides, amino acids and amines (50). Dietary nitrates can also be reduced to nitrites by the formation of nitric oxide when inflammation is present (32, 49, 50). Increased GC risk has been found with nitrosamines formation when H. pylori infection or decrease vitamin C plasma levels are present (51). Different experimental and observational studies suggest that nitrosamine and consumption of processed foods with related substances increase the GC risk (32).
Alcohol. The association of alcohol with GC is controversial. Available data do not support the concept that this substance is associated with increased GC risk (32)
Helicobacter pylori
H. pylori was the first bacterial pathogen to be classified as a type I carcinogen by the International Agency for Research on Cancer (52). This rating was originally based on the results of three large epidemiological studies (53-55) conducted before the association was demonstrated experimentally (56, 57). This pronouncement by the IARC, without experimental evidence, was criticized by some scientists as hasty. The epidemiological studies mentioned showed that the GC risk for people infected with the bacteria ranged between 2.8 and 6 times greater than uninfected people. The EUROGAST international study also found that those infected had an increased GC risk with an OR of 6 (58) A relatively recent metaanalysis found that the risk was 2.28 for individuals with GG whose serology was positive for H. pylori and 2.87 when the microorganism was Cag A (+) (59). The studies mentioned above, and other more recent studies, have concluded that the evidence on the relationship between H. pylori and GC is unequivocal. This microorganism is the main causative agent of this tumor (60-63), however a small proportion of GCs may possibly be associated with Epstein Barr virus (64).
It is currently believed that H. pylori is responsible for more than 90% of GCs (62). Nevertheless, H. pylori infection is not sufficient to cause gastric cancer as only a minority of those infected will develop GC. Today the causal association of this infection is no longer disputed. The controversial areas now lie in the identification of the mechanisms by the bacteria produce GC and in methods for identification of individuals at high risk of developing GC.
All H. pylori are pathogens and cause chronic gastritis in all infected persons (62, 65-68). However, some organisms are more virulent than others. Chronic gastritis is asymptomatic. Different types are associated with different final clinical outcomes: antral gastritis with duodenal ulcer cases and body pangastritis/gastritis with gastric cancer or gastric ulcer cases (68-71) as shown in figure 1. The associations between these two types of gastritis and these two diseases has been recognized for over 70 years (72, 73).
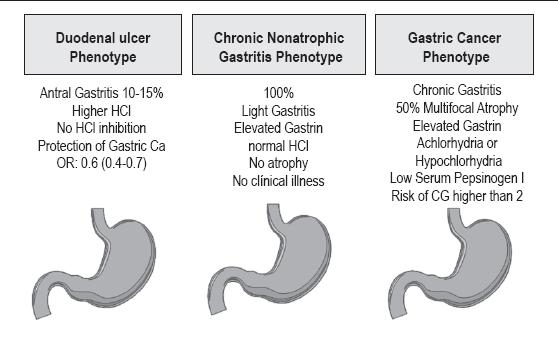
Figure 1. Patterns of gastritis associated with H. pylori. Modified from ref. 108. Amieva MR, El-Omar. Gastroenterology 2008; 134: 306-23.
The ultimate consequences of the infection depend on genetic characteristics of the host, on the characteristics of the particular type H. pylori and on external environmental factors. The two types of chronic gastritis produce different alterations in gastric physiology. In antral gastritis, there is acid hypersecretion (70, 71) which results in duodenal bulb gastric metaplasia with alteration of defense mechanisms and eventual duodenal ulcer. It has long been known that patients with duodenal ulcers, despite having antral gastritis, have reduced GC risk (62, 69, 74), whereas gastric ulcers increase the GC risk (75). In Japan, where incidence and prevalence of GC is high, the duodenal ulcer/gastric cancer ratio is less than 1 (76). In regions with low GC prevalence, including some Western countries and Southeast Asia, the ratio is greater than 1 (77, 78). In Japan Uemura et al (74) found that none of the patients infected with H. pylori who had duodenal ulcers developed gastric cancer during the eight years they were tracked. In contrast, 3.4% of the patients with gastric ulcers developed GC. That study also confirmed that patients with duodenal ulcers and antral gastritis usually had little or no atrophy, whereas those with gastric ulcers usually had body-antral gastritis with various degrees of atrophy. The predominant body gastritis was associated with a relative risk (RR) of 34.5 for GC (74).
The mechanisms by which H. pylori produces GC are not fully known, but today carcinogenesis may involve indirect mechanisms (permanent inflammation) and direct mechanisms represented by the action of different virulence factors of H. pylori on the gastric epithelium (76).
INDIRECT MECHANISMS
These are related to the B inflammatory response produced in the infected stomach. It causes morphological and molecular changes in the epithelium resulting in the following sequence. Histopathologically, in 1-2% of those infected, chronic gastritis develops, followed by gastric atrophy, complete intestinal metaplasia, incomplete intestinal metaplasia, dysplasia and cancer (68, 79, 80). Atrophy, which is usually present, may have a multifocal or diffuse pattern and may be associated with a form of metaplastic mucosa called pseudopyloric (body antralization) (81). This is also known as metaplasia expressing spasmolytic polypeptide, since these antral gastric body glands express this polypeptide which is a trefoil peptide normally present in the intestinal mucosa in normal and dysplastic cells and in neoplastic cells (82).
These hypothetical pathways suggest that chronic inflammation leads to gastric atrophy, a preneoplastic stomach lesion. Chronic inflammation causes an increase in tissue turnover with excessive cell proliferation. This can predispose to frequent mitotic errors with increased risk of mutagenesis (69, 76, 80, 82). The concurrence of cellular hyperproliferation with inflammation involves the generation cytokines, growth factors and free radicals of oxygen and nitrogen (nitric oxide) (62, 68, 83). This favors the possibility of damage to the DNA of the gastric cells which may induce mutations in the DNA, or genes, "silencing" them at the transcription level (62, 68). Matsumoto et al found that H. pylori infection caused induced activation expression of the cytidine deaminase (AID) gene (84). This can predispose the p53 tumor suppressor gene to mutations (85). Harmful environmental factors such as smoking and high levels of salt in the diet further increase the GC risk, whereas diets high in antioxidants such as fresh fruit and vegetables may be protective (31).
This sequence of events is the classic paradigm of Professor Pelayo Correa's model (12, 26, 68). Dr. Correa has the merit of having proposed the theory of gastric carcinogenesis over 30 years ago prior to the discovery of H. pylori. The process involves a multi-step progression from gastritis to cancer (86). Dr. Correa's paradigm gradually became more important with the discovery of H. pylori as the main cause of chronic gastritis (87) (figure 2). This model has continued to evolve a number of changes that affect the natural history of infection have been identified (82). Correa posited that atrophic gastritis was multifocal, occurred throughout the whole stomach and was more frequent in geographical areas with the highest incidences of GC (88, 89). In addition, he demonstrated that populations with higher risks of GC in Colombia had higher prevalences of atrophic gastritis than those with lower risks for the tumor. This confirmed observations made previously by other authors (90, 91). Although Dr. Correa's model of carcinogenesis posits the start of CG with chronic gastritis which later progresses to atrophy and then to intestinal metaplasia-dysplasia-cancer in a sequential manner, whether or not atrophy precedes metaplasia remains unknown. The two could occur simultaneously. Verification of this sequence would only be possible if individual lesions could be followed prospectively without any intervention (92).
The concept of association between inflammation and GC had already been recognized by Virchow in 1863, when he hypothesized that cancer originates in sites with chronic inflammation (93).
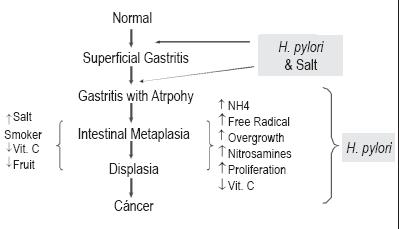
Figure 2. Pelayo Correa's model of carcinogenesis .
H. pylori infection stimulates both innate and acquired immune responses (94). The initial step in this process is recognition of the microorganism through Nod1 (nucleotide-binding oligomerization domain protein I) which is an innate system for detecting bacteria which identify a Gram negative peptidoglycan muropeptides of these bacteria (94-97).
Immune system stimulation after H. pylori recognition by Nod1 results in chronic gastritis. The infiltrating inflammatory cells colonize the epithelium which influences colonization density, inflammation level and generation of adaptive immune response (94, 95). In this way the innate response is a key determinant of the severity of the disease and gastric carcinogenesis. This innate immune response against H. pylori, includes the release of antibacterial peptides and infiltration of the mucosa by all types of immune effector cells. Improper recognition of H. pylori by the innate immune system may contribute to the failure of the adaptive immune system to eliminate it. Certain polymorphisms of Toll-Like Receptor 4 3725 G/C have been found associated with increased risk of gastric atrophy (99) as well as GC (100). This indicates that this variation of innate immune system transmembrane protein recognizes pathogenic molecular patterns. Thus they are host factors involved in the response and outcome of infection. Besides the primary response, there is also an acquired humoral, local and systemic cellular immune response which persists throughout life. The response of the T cell is primarily Th1 (96, 97, 101, 102). This is the "wrong" answer, since H. pylori is an extracellular germ which, like similar microorganisms, should trigger a Th2 response (96, 97, 101, 102). The Th1 response produces interferon (IFN g), a tumor necrosis factor alpha (TNF a). IL-12, IL-18 (94-97). The polarization of the immune response towards Th1 with its cytokine profile may contribute to the development of more severe gastric pathology. In contrast, the activation of a Th2 response and the expression of cytokines such as IL-4, produce decreased gastric inflammation and protect against more severe pathologies, probably including GC. They counteract the effects of Th1 cytokines (96, 101, 102). These type I cytokines activate macrophages which in turn secrete proinflammatory factors along with adding bactericidal capacity, compared to activation by Th2 cell response (95-97). The severity of chronic gastritis is correlated with the number of cells that secrete IFN g (96, 62, 101, 102). The immune response differentiation towards Th1 seems to be influenced by the bacteria themselves and by environmental factors (62, 68, 96, 101). Parasitic infections which induce Th2 response have been suggested as one factor explaining the lower incidence of gastric cancer in regions of Africa with high prevalence of H. pylori infection (the "African enigma") (103). However, this "enigma" has been challenged by some experts who believe it is really a myth since duodenal ulcers and GCs are both present in Africa (73). It was recently found that IL-17 and IL-23 are involved in H. pylori infection (96). The IL-17 cytokines produced by Th17 lymphocytes alter the immune surveillance and promote tumor growth (96). In China, Zhang et al (104) found that IL-23 and IL-17 were significantly elevated in patients with advanced GC, suggesting that Th17 cells may contribute to the carcinogenesis promoted by H. pylori.
When a person is infected with H. pylori, the GC risk is 2 to 3 times greater than for uninfected people, but if you have anti Cag A antibodies the risk increases to 11 times greater. Furthermore, depending on the degree of protein phosphorylation, this risk may increase further (105). If to all this is added the alteration of the gene that encodes synthesis of IL-1B-511, the risk increases to 87 times greater (68) (table 1). Modulation of the inflammatory process largely determines whether the outcome becomes neoplastic or not (60, 62, 68, 79, 70). A major difference between these two possibilities is acid secretion which, as mentioned, is normal in patients who have DU and increased in patients with gastric ulcer. Patients with body-antral gastritis generally have acid hyposecretion (62, 67, 70, 79). In the latter type of gastritis, host factors such as IL-1, IL-8 and matrix metalloproteinases, together with virulence factors of H. pylori protein Cag A, VacA and OipA, play an important role in the development of gastric ulcers (60, 107-110).
Table 1. H. pylori and gastric cancer risk.

Gastric hyposecretion is partly determined by interleukin 1B (IL-1B). Besides being a proinflammatory cytokine, it is a potent inhibitor of acid secretion (100 times more potent than omeprazole) (111). Some investigators have found that polymorphisms of IL-1B gene and the antagonist of IL-1 (IL-1RN) are associated with both hypochlorhydria and increased GC risk (112-114), but others have not found any association with CG (115-17). Three metaanalyses have been conducted regarding this controversy. They have included multiple publications on the subject including analysis of IL-1B and IL-1RN polymorphisms (118-120). Two of them concluded that proinflammatory interleukin increases the risk of GC (118, 119), while the third concluded that it did not (120). The cause of this contradiction is not known, although, given the multifactorial nature of GC it is conceivable that other genetic susceptibility factors, lifestyle factors and environmental factors may mitigate the effect of this particular polymorphism.
Despite the spread and growing support for the multiple step gastric carcinogenesis hypothesis, for some it is still questionable whether or not metaplasia is a premalignant condition. One reason for this is that diffuse gastric carcinomas, like signet ring carcinoma, do not appear to be associated with either atrophy or intestinal metaplasia. For this reason some experts think that these should be defined as paraneoplastic disorders instead of preneoplastic (121). However, more than a decade ago it was shown that intestinal types I and II metaplasia have no GC risks, but that type III has a relative risk of 4.6 (122). Furthermore, an 8 to 9 year follow-up study of 90 patients with type III intestinal metaplasia found that only one patient developed CG (123). Another more recent study by El-Zimaity et al (124), obtained similar results. They found no dysplasia or carcinoma in any of 33 patients with type II metaplasia and 34 patients with type III metaplasia during a 9 year follow-up. In Japan Kakinoki et al (125) recently found that the majority of gastric adenocarcinomas in patients infected with H. pylori developed when mild to moderate atrophy existed. These latest studies keep the controversy over the role of gastric metaplasia as a precursor of GC alive. Now there is evidence that preneoplastic and neoplastic cells, bone marrow derived progenitor cells (homing stem cells) in adults, have contributed to the chronic stomach inflammation induced and perpetuated by H. pylori (126-128). These new findings do not discredit Correa's model, but rather enrich it and highlight its importance. Different inflammatory mediators are key to mobilizing the bone marrow derived progenitor cells and modulating the GC risk (figure 3). Atrophy and metaplasia are permanent chronic inflammation indicators which are crucial for GC's production of the three states of carcinogenesis: initiation, promotion and progression (126-128). Inflammation induced by H. pylori involves complex molecular networks which are only partially known. They generate constant tissue destruction culminating in atrophy and intestinal metaplasia in genetically susceptible individuals when favorable environmental conditions exist. Initial repair is undertaken without success by stem cells from the peripheral blood. They are progressively replaced by bone marrow derived progenitor cells, the second line of defense, before the onset of severe ongoing inflammation and tissue destruction (82). Similarly, bone marrow derived progenitor cells could be the stem cell of gastric cancer. Given its high degree of plasticity it can give rise to many vary in different types of cells including those that form different structures of the tumor (129).
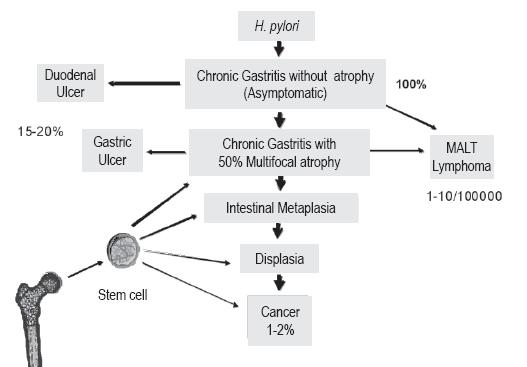
Figure 3. Outcomes of H. pylori infections.
H. PYLORI DIRECT MECHANISMS (VIRULENCE FACTORS)
H. pylori is genetically more diverse than most other bacterial populations (130). This genetic diversity is considered to be a feature involved in this microorganism's ability to produce different diseases. Since it is beyond the scope of this work to discuss the many virulence factors of this organism, only some which we consider to be the most relevant will be highlighted.
The cag A Pathogenicity island (cag PAI)
Most strains of H. pylori can be grouped into two distinct phenotypes based on the presence of the cag PAI (108, 110, 130, 131). The cag PAI is a new region of DNA acquired by H. pylori in the course of its evolution by horizontal transfer from other bacteria (131). This island contains 30 to 40 genes including the cytotoxin associated cag A, which encodes the Cag A protein (108, 130, 131). The cag PAI is present in 60% of H. pylori in Western countries and in more than 90% of the countries of Eastern Asia (107, 132). Cag A protein is highly immunogenic. It has a molecular weight between 128 and140 kDa. The strains expressing this protein are more virulent than those that do not express it. (110,130-132). The first induce increased production of inflammatory cytokines such as IL-8, increased cellular proliferation and apoptosis (62, 107, 133). H. pylori injects these Cag A proteins into the cytoplasm of the epithelial cell by a Type IV secretory mechanism (molecular syringe) (134-136). Translocation of Cag A into gastric epithelial cells produce significant structural and functional changes that benefit bacteria (134). Once injected, the cell recognizes the protein as an epithelial signaling molecule. Similar to other signaling protein it is phosphorylated in varying degrees at sites that contain the Glu-Pro-Ile-Tyr-Ala sequence of five amino acids called EPIYA motifs (130, 134-136). When CagA is phosphorylated by these kinases it activates the tyrosine phosphatase SHP-2 Oncoproteins whose mutation is associated with malignancy in humans (130-136, 137.138). CagA deregulates SHP-2 to disrupt the Erk MAP kinase and FAK dephosphorylating focal adhesion kinases involved in inducing changes in cell morphology resulting in a highly elongated cell morphology termed the hummingbird phenotype (135-137). Cag A also damages cell-cell interactions independently of phosphorylation, destroys closed joints and causes loss of polarity in epithelial cells. It also destabilizes the E-cadherin / β-catenin complex (22, 130). Recently, the first experimental evidence that Cag A is real H. pylori Oncoprotein was obtained by demonstrating that in transgenic mice phosphorylation of Cag A is associated with tumors but not when it inhibited the ability to phosphorylate (139).
The higher the phosphorylation levels of Cag A protein the greater the potential for an oncogenic strain of H. pylori. Depending on phosphorylation site (EPIYA reasons), the Cag A protein may be one of two sub-types (130.139): Eastern Asian Cag A and Western CagA (130, 135). The EPIYA motif is part of four different EPIYA sites: EPIYA-A,-B,-C and-D (138.139). Each site is determined by the amino acid sequence surrounding the sequence of EPIYA. In Western countries, the most common are Cag A EPIYA-A and B, followed by C sites, where C is repeated from one to three times (ABC, ABCC and ABCCC). The most common type is the ABC variant (135). In Asian countries most of the Cag A is EPIYA-A and-D (ABD type). The importance of identifying these different types of Cag A lies in the different ways they are associated with GC prevalence and mortality. The Cag A of various regions of the world with high prevalence rates of GC are similar to East Asian Cag A. In contrast, where there is low prevalence of gastric cancer CagA is the Western type (130, 140).
In addition to the Cag A proteins, small effector molecules such as cell wall peptidoglycan are released into gastric cells by the type IV secretion system (141). In cell cytoplasm peptidoglycan is recognized by the innate immune system defense protein NOD1, leading to activation of nuclear factor k beta (NF-kB) which increases the expression of genes encoding pro-inflammatory proteins like IL -8, CXC chemokines and defensin B, an antimicrobial peptide (141).
Gene vacA and protein VacA
VacA or vacuolating cytotoxin is a protein. It is one of the most important virulence factors of H. pylori. It is encoded by the gene vacA. It has a molecular weight of approximately 139 kDa and derives its name from one of its studied early effects, the production of massive vacuolization of gastric epithelial cells (110, 142,143-146). Unlike cagA, all strains of H. pylori have vacA. However, its functional expression varies, so not all induce vacuolation of epithelial cells (110, 142, 143). While this gene is present in all strains of H. pylori, has several polymorphisms (142, 143). Typically, it has been considered to have two regions: the "s" region or signal sequence region and the genome media region or "m". For each there are two alleles: s1 and s2, and m1 and m2. All possible combinations of the two occur: s1m1, s1m2, s2m2 etc. The s1 and m1 in turn can be subdivided into s1a, s1b, s1c, m1a, m1b and m1c (143-145). Besides cellular vacuolization, VacA produces alterations in the pore formation of the gastric epithelium (110, 147), in alteration of close intercellular unions, in apoptosis, in suppression of the host's immune system, in blocking macrophage phagosomes, in interference with antigen presentation to T cells, and in the inhibition of activation and proliferation of T lymphocytes (down regulation) (110, 146). The genotype s2 blocks vacuolating activity while vacA s1 strains are more cytotoxic and are associated with increased gastric inflammation and duodenal ulcers (146). In general vacA s1m1 strains produce larger amounts of cytotoxin and are associated with more severe gastritis, atrophy, intestinal metaplasia, and gastroduodenal ulcers (110,143-146). In contrast vacA s2m2 genotype strains are less virulent and produce little or no cytotoxin. They induce milder gastritis and are associated with less CG (110.143, 146). Sugimoto recently described strains with genotypes s1m1 having higher risks of GC in Latin American countries (145). For gastric cancer vacA s1 has an OR of 4.17, while vacA m1 has an OR of 3.6. In addition to these classic vacA polymorphisms a new region of the gene has recently been described. It is called the intermediate region (i) which in turn has two sub-regions: i1 and i2 (147). Previous studies had found that in patients with GC s1m1strains were found most frequently, while s1m2 strains were occasionally found (145.146). In this study, we found that all s1m1 and s1m2 strains with in vitro vacuolating activity were included in the intermediate region i1. For this reason the authors have concluded that this new genotype would be the best carcinogenic marker for vacA (147).
Other virulence factors
Besides the vacA and cagA H. pylori has many other virulence factors. Among them are the inflammatory outer membrane protein (OMP), Blood Group Antigen Binding Adhesin (BabA) and AlpAB adhesin (60, 107.110, 148 -151). BabA, is a foreign protein which allows H. pylori to adhere to Lewis B antigens expressed on the membranes of gastric epithelial cells (148). Strains expressing these proteins produce more severe lesions. The respective proteins encoded by these genes are closely related to H. pylori associated gastroduodenal diseases, including GC (73, 140).
So far, no one knows the exact mechanism of action of H. pylori's multiple virulence factors. Knowledge of them, their interactions and their genetic variation could help identify patients at increased GC risk (59, 73, 80). For example the interaction of CagA positive strains with the 511 polymorphism of IL-1B l increases the risk of GC, while association of inflammatory OMP with cagPAI influences the levels of IL-8 since both are needed to activate the promoter region of these cytokines (150, 151). Other additional mechanisms by which H. pylori promotes carcinogenesis (152) are shown in table 2.
Table 2. Oncogenic mechanisms produced by H. pylori.

Host genetic factors
Genetic polymorphisms of the infected individual influence the various clinical manifestations of H. pylori infections. Among the previously mentioned polymorphisms, IL-1B and IL-1RN are associated with increased GC risk (118-120). Another independent GC risk factor is tumor necrosis factor alpha (TNFa) genotypes (153). TNFa is a cytokine with B proinflammatory activity. It is also induced by H. pylori (8, 60, 93, 96, 108) Like IL-1B, but to a lesser extent, it also inhibits the production of HCl.
The A allele of this cytokine has an OR of 2.2 for CG (95% CI 1.4-3.7) (154). In contrast, IL-10 is an anti-inflammatory cytokine that produces negative regulation (down regulation) of proinflammatory cytokines (IL-1B, TNFa, interferon G etc). Its deficiency contributes to increasing the Th1 immune response, and with it increased gastric inflammation (80, 96, 152, 153). Other described polymorphisms that are associated with increased risk of GC are those of the IL-8 promoter region (155, 156).
Gastric cancer prevention
Taking into account the incontrovertible association of H. pylori with gastric cancer, eradication of these bacteria should be an excellent strategy for preventing this cancer. However, the efficacy of this strategy has yet to be demonstrated. Currently it is agreed that the optimum moment for eliminating the microorganism and diminishing the risk of gastric cancer is prior to the development of atrophy and intestinal metaplasia (157-159). This was demonstrated by Wong in China (158). Recently a case was reported of two patients in whom H. pylori was eradicated after intestinal metaplasia and atrophy had developed who later developed gastric cancer (160). For this reason the development of after intestinal metaplasia and atrophy can be considered as the point of no return for the development of gastric cancer. Eradication of the bacteria after this point does not diminish the risk of gastric cancer (158-160). The implication is that the eradication of H. pylori does not prevent gastric cancer in all patients, and when these advanced gastric lesions exist, endoscopic follow up is necessary for early detection of gastric cancer (160). On the other hand, for patients with gastric MALT lymphoma, eradication of H. pylori cures the lymphoma in the majority of patients (161).
Recently a transcendental advance in the prevention of H. pylori was achieved in a double blind phase I study undertaken by Malfertheiner and colleagues (162). They found a prophylactic recombinant vaccine which uses VacA, CagA and the neutrophil activating protein of H. pylori (NAP-HP) to induce antibody production. The vaccine was successful in 86% of the healthy volunteers who participated in the study. Their ages were between 18 and 40. The authors' conclusion is that this combination of antigens has an acceptable level of safety and immunogenicity and stimulates memory T and merits additional clinical study.
Conclusions
There are two pathways involved in H. pylori carcinogenesis. One is through indirect mechanisms of inflammation induced by persistent infection accompanied by hyper proliferation of cells with a high risk of DNA damage by free radicals. Bone marrow derived stem cells are part of this process and could be the stem cells of gastric cancer. The second route involves direct actions of H. pylori proteins on gastric cells. In the light of available information it is likely that both pathways are involved in the genesis of gastric cancer (figure 4). Among the host's genetic factors there is evidence that genetic polymorphisms of IL-1B, TNF, IL-8, gama INF and IL-10 among others induce B inflammatory responses that are associated with increased GC risk. Finally gastric cancer is a multifactorial disease involving genetic factors of the individual, environmental factors and most importantly, H. pylori infection (figure 5).

Figure 4. H. pylori and carcinogenesis pathways.
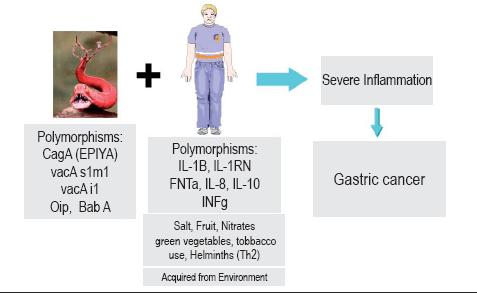
Figura 5. H. pylori and gastric cancer (agent, host, environment).
REFERENCES
1. Ferlay J, Bray F, Pisani P, et al. GLOBOCAN 2002: Cancer Incidence, Mortality and Prevalence Worldwide, versión 2.0 IARC CancerBase No 5 Lyon: IARC 2004.
2. Parkin DM, Bray F, Ferlay J, Paisani P. Global Cancer Statistics 2005; CA Cancer J Clin 2005; 55: 74-108.
3. Krew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354-62.
4. Hamashima C, Shibuya D, Yamazaki H, Inoue K, Fukao A, Saito Sobue T. The Japanese guidelines for gastric cancer screening. Jpn J Clin Oncol 2008; 38: 259-67.
5. Piñeros M, Hernández G, Bray F. Increasing mortality rates of common malignancies in Colombia. Cancer 2004; 101: 2285-92.
6. Correa P, Piazzuelo MB, Camargo MC. Overview and pathology of gastric cancer. En Wang T, Fox J, Giraud A. (Edit). The biology of gastric cancers. Springer Science Business + Media 2009. p. 21-44.
7. Smith MG, Hold GL, Tahara E, El-Omar EM. Cellular and molecular aspects of gastric cancer. World J Gastroenterol 2006; 12: 2979-90.
8. Mosss SF, Malfertheiner P. Helicobacter and gastric malignancies. Helicobacter 2007; 12(Suppl 1): 23-30.
9. Farinha P, Gascoyne RD. Helicobacter pylori and MALT lymphoma. Gastroenterology 2005; 128: 1579-1605.
10. Jarvi O, Lauren P. On the role of heterotopias of the intestinal epithelium in the pathogenesis of gastric cancer. Acta Pathol Microbiol Scand 1951; 29: 26-44.
11. Lauren, P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31-49.
12. Correa P, Carneiro F. Classification of gastric carcinomas. Curr Diagn Pathol 1997; 4: 51-9.
13. Ekstrom AM, Serafini M, Nyren O, Hansson LE, Ye W, Wolk A. Dietary antioxidante intake and the risk of noncardia cancer for the intestinal and diffuse types: a population-based case-control study in Sweden. Int J Cancer 2000; 87: 133-40.
14. El-Omar EM, Lochhead P. Gastric cancer. Br Med Bull 2008; 85: 87-100.
15. Henson DE, Dittus C, Younes M, Nagyen H, Albores-Saavedra J. Differential trends in the intestinal and diffuse topless of gastric carcinoma in the United States-2000: increase in the signet ring cell type. Arch Pathol Lab Med 2004; 128: 765-70.
16. Oliveira C, Seruca R, Carneiro F. Hereditary gastric cancer. Best Pract Res Clin Gastroenterol 2009; 23: 147-57.
17. Alberts SR, Cervantes A, van de Velde CJ. Gastric cancer: epidemiology, pathology and treatment. Ann Oncol 2003; 14 (suppl 2): ii31-ii36.
18. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392: 402-5.
19. Carneiro F, Sobrinho-Simoes M. Hereditary diffuse gastric cancer: lessons from histopatholgy. Adv Anat Pathol 2005; 12: 151-2.
20. Medina-Franco H, Barreto-Zúñiga R, García-Álvarez MN. Preemptive total gastrectomy for hereditary gastric cancer. J Gastrointest Surg 2007; 11: 314-7.
21. Medina-Franco H, Medina AR, Vizcaíno G, Medina-Franco JL. Single nucleotide polymorphisms in the promoter region of the E-cadherin gene in gastric cancer: case-control study in a young Mexican population. Ann Surg Oncol 2007; 14: 2246-9.
22. Cisco RM, Ford JM, Norton JA. Hereditary diffuse gastric cancer. Cancer 2008; 113 (7 Suppl): 1850-6.
23. Masciari S, Larsson N, Senz J, Boid N, Kaurah P, Kandel MJ, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet 2007; 44: 726-31.
24. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19 (Suppl.1): S37-S43.
25. Kabir S. Effect of Helicobacter pylori eradication on incident of gastric cancer in human and animal models: underlying biochemical and molecular events. Helicobacter 2009; 14: 159-71.
26. Correa P, Chen VW. Gastric Cancer. Cancer Surv 1994; 19: 55-76.
27. Kono S, Irohata T. Nutrition and stomach cancer. Cancer Causes Control 1996; 7: 41-55.
28. Larsson SC, Bergkvist L, Wolk A. Fruit and vegetable consumption and incidence of gastric cancer: A prospective study. Cancer Epidemiol Biomarkers Prev 2006; 15: 1998-2001.
29. Bae JM, Lee EJ, Guyatt G. Citrus fruit intake and stomach cancer risk: a quantitative systematic review. Gastric Cancer 2008; 11: 23-37.
30. González CA, Pera G, Agudo A, Bueno-de Mezquita HB, Ceroti M, et al. Fruit and vegetable intake and the risk of stomach and oesophagus adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC-Eurogast). Int J Cancer 2006; 118: 2559-66.
31. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for preventing gastrointestinal cancers. Cochrane Database of Systematic Reviews 2004; Issue 4.
32. Liu C, Huang XD, Russell RM. Diet and gastric cancer. En Wang TC, Fox J, Giraud A. (Edits). The biology of gastric cancers. Springer Science+Business Media LLC 2009. p. 59-89.
33. Correa P, Fontham ET, Bravo JC, Bravo LE, Ruiz B, Zarama G, Realpe JL, Malcom GT, Li D, Johnson WD, Mera R. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-helicobacter pylori therapy. J Natl Cancer Inst 2000; 92: 1881-8.
34. Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005; 54: 1536-40.
35. Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ, Yang CS, Zheng SF, Gail M, Li GY, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst 1993; 85: 1483-92.
36. Goswami UC, Sharma N. Efficiency of a few retinoids and carotenoids in vivo in controlling benzo[a]pyrene-induced forestomach tumour in female Swiss mice. Br J Nutr 2005; 94: 540-3.
37. Velmurugan B, Bhuvaneswari V, Burra UK, Nagini S. Prevention of N-methyl-N';-nitro-N-nitrosoguanidine and saturated sodium chloride-induced gastric carcinogenesis in Wistar rats by lycopene. Eur J Cancer Prev 2002; 11: 19-26.
38. Velmurugan B, Bhuvaneswari V, Nagini S. Antiperoxidative effects of lycopene during N-methyl-N';-nitro-N-nitrosoguanidine-induced gastric carcinogenesis. Fitoterapia 2002; 73: 604-11.
39. Velmurugan B, Mani A, Nagini S. Combination of S-allylcysteine and lycopene induces apoptosis by modulating Bcl-2, Bax, Bim and caspases during experimental gastric carcinogenesis. Eur J Cancer Prev 2005; 14: 387-93.
40. Liu C, Russell RM, Wang XD. Lycopene supplementation prevents smoke-induced changes in p53, p53 phosphorylation, cell proliferation, and apoptosis in the gastric mucosa of ferrets. J Nutr 2006; 136: 106-11.
41. Wang X, Willen R, Wadstrom T. Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob Agents Chemother 2000; 44: 2452-7.
42. Liu BH, Lee YK. Effect of total secondary carotenoids extracts from Chlorococcum sp on Helicobacter pylori-infected BALB/c mice. Int Immunopharmacol 2003; 3: 979-86.
43. World Cancer Research Fund, American Institute for Cancer Research. Food, nutrition and the prevention of cancer: a global perspective. Washington, DC: American Institute for cancer Research 1997.
44. Kato S, Tsukamoto T, Mizoshita T, Tanaka H, Kumagai T, Ota H, Katsuyama T, Asaka M, Tatematsu M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer 2006; 119: 1558-66.
45. Watanabe H, Takahashi T, Okamoto T, Ogundigie PO, Ito A. Effects of sodium chloride and ethanol on stomach tumorigenesis in ACI rats treated with N-methyl-N';-nitro-Nnitrosoguanidine: a quantitative morphometric approach. Jpn J Cancer Res 1992; 83: 588-93.
46. Furihata C, Sato Y, Hosaka M, Matsushima T, Furukawa F, Takahashi M. NaCl induced ornithine decarboxylase and DNA synthesis in rat stomach mucosa. Biochem Biophys Res Commun 1984; 121: 1027-32.
47. Sorbye H, Kvinnsland S, Svanes K. Effect of salt-induced mucosal damage and healing on penetration of N-methyl-N';-nitro-N-nitrosoguanidine to proliferative cells in the gastric mucosa of rats. Carcinogenesis 1994; 15: 673-9.
48. Fox JG, Dangler CA, Taylor NS, King A, Koh TJ, Wang TC. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res 1999; 59: 4823-8.
49. Jakszyn P, González, CA. Nitrosamine and related food intake and gastric and oesophageal cancer risk: a systematic review of the epidemiological evidence. World J Gastroenterol 2006; 12: 4296-303.
50. Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett.1995; 93: 17-48.
51. Jakszyn P, Bingham S, Pera G, Agudo A, Luben R, Welch A, et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis.2006; 27: 1497-501.
52. Schistosomes, Liver fluyes and Helicobacter pylori IARC Working group on the evaluation of carcinogenic risks to humans. Lyon, 7-14 June 1994. IARC Monog Eval Carcinog Risks Hum 1994; 61: 1-41.
53. Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, et al. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 1991, 302: 1302-5.
54. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentrich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127-31.
55. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser M, et al. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132-6.
56. Sugiyama A, Maruta F, Ikeno T, Ishida K, Kawasaki S, Katsuyama T, et al. Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 1998; 58: 2067-69.
57. Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998; 115: 642-8.
58. An International association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993; 341: 1359-62.
59. Huang JQ, Zheng GF, Sumanak K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cag A seropositivity and gastric cancer. Gastroenterology 2003; 125: 1636-44.
60. Peter S, Begglinnger C. Helicobacter pylori and gastric cancer: The causal relationship. Digestion 2007; 75: 25-35.
61. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med 2002; 347: 1175-86.
62. Mueller A, Falkow S, Amieva MR. Helicobacter pylori and gastric cancer: what can be learned by studying the response of gastric epithelial cells to the infection? Cancer Epidemiol Biomarkers Prev 2005; 14: 1859-64.
63. Peter S, Beglinger C. Helicobacter pylori and gastric cancer: the causal relationship. Digestion 2007; 75: 25-35.
64. Wu MS, Shun CT, Wu CC, et al. Epstein Barr virus associated with Helicobacter pylori. Infection and genetic alterations. Gastroenterology 2000; 118: 1031-8.
65. Report of the Digestive Health Initiative International Update. Conference on Helicobacter pylori. Gastroenterology 1997; 113 (Suppl): S4-S8.
66. Saad R, Chey W. A clinician's guide to managing Helicobacter pylori infection. Clev Clin J Med 2005; 72: 109-124.
67. Parsonnet J. Helicobacter pylori: the size of the problem. Gut 1998; 43: S6-S9.
68. Correa P, Schneider BG. Etiology of gastric cancer: what is new? Cancer Epidemiol Biomarkers Prev 2005; 14: 1865-8.
69. Guillen D, McColl KEL. Gastroduodenal disease, Helicobacter pylori, and genetic polymorphisms. Clinical Gastroenterol Hepatol 2005; 3: 1180-86.
70. El Omar EM, Penman I, Ardill JE, Chittajallu RS, Howie C, Mc Coll KE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995; 109:681-91.
71. Lai LH, Sung JJY. Helicobacter pylori and benign upper digestive disease. Best Pract Res Clin Gastroenterol 2007; 21: 261-79.
72. Faber K. Chronic gastritis: its relation to achylia and ulcer. Lancet 1927; 2: 902-7.
73. Graham DY, Lu H, Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: facts or medical myths. J Dig Dis 2009; 10: 77-84.
74. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784-9.
75. Chiba T, Seno H, Marusawa Y, Okazaki K. Host factors are important in determining clinical outcomes of Helicobacter pylori infection. J Gastroenterol 2006; 41: 1-9.
76. Chiba T, Marusawa H, Seno H, Watanabe N. Mechanism Fort he gastric cancer development by Helicobacter infection. J Gastroenterol Hepatol 2008; 23: 1175-81.
77. Bonnevie O. The incidence of duodenal ulcer in Copenhagen County. Gastroenterology 1975; 10: 385-93.
78. Hu PJ, Li YY, Zhou MH, Chen MH, Huang BJ, Mitchel HM, et al. Helicobacter pylori associated with a high prevalence of duodenal ulcer disease and low prevalence of gastric cancer in a developing nation. Gut 1995; 36: 198-202.
79. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg pathol 1995; 19: S37-S43.
80. Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori Basic research to patient care. Gastroenterology 2006; 130: 188-206.
81. Matsukura N, Suzuki K, Kavachi T, Aoyagi M, Sugimura T, Kitaoka H, et al. Distribution of marker enzymes and mucin in intestinal metaplasia in human stomach and relation to complete and incomplete types of intestinal metaplasia to minute gastric carcinomas. J Natl Cancer Inst 1980; 65: 231-40.
82. Correa P, Hoghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007; 133: 659-72.
83. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 195-200.
84. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T. Helicobacter pylori infection triggers aberrant expresión of activation-induced cytidine deaminase in gastric epithelium. Nat Med 2007; 13: 470-6.
85. Takaishi S, Wang TC. Providing AID to p53 mutagenesis. Nat Med 2007; 13: 404-6.
86. Correa P, Haenszel W, Cuello C, Tannembaum S, Archer M. A model for gastric cancer epidemiology. Lancet 1975; 2: 58-60.
87. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 1273-5.
88. Correa P. The epidemiology and pathogenesis of chronic: three etiologic entities. Front Gastrointest Res 1980; 6: 98-108.
89. Correa P. Clinical implications of recent development in gastric cancer pathology and epidemiology. Sem Oncol 1985; 12: 2-10.
90. Bonne C, Hartz PH, Klerks JV, Posthuma JH, Radsma W, Tjokronegoro S, et al. Morphology of the stomach and gastric secretion in Malays and Chinese and the different incident of gastric ulcer and cancer in these races. Am J Cancer 1938; 33: 265-79.
91. Siurala M, Varis K, Wiljasalo M. Studies of patients with atrophic gastritis: a 10 -15 year follow-up. Scand J Gastroenterol 1966; 1: 40-8.
92. Volk J, Parsonnet J. Epidemiology of Gastric Cancer. En Wang TC, Fox J, Giraud A. (Eds). The biology of Gastric cancers. Springer Science+ Business Media LLC 2009. p. 25-57.
93. Matysiak-Budnik T, Mégraud F. Helicobacter pylori infection and gastric cancer. Eur J Cancer 2006; 42: 708-16.
94. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. NodI detect a unique muropeptide from gram negative bacterial peptidoglycan. Science 2003; 300: 1584-87.
95. Girardin SE Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, et al. Peptiglycan molecular requeriments allowing detection by NodI and Nod2. J Biol Chem 2003; 278: 41702-8.
96. Keeffe J, Moran AP. Conventional, regulatory and unconventional T cells in the immunologic response to Helicobacter pylori. Helicobacter 2008; 13: 1-19.
97. Ma J, Chen T, Mandelin J, Ceponis A, Miller NE. Regulation of macrophage activation. Cell Mol Life Sci 2003; 60: 2334-46.
98. Malaty HM. Epidemiology of Helicobacter pylori. Best Pract Res Clin Gastroenterol 2007; 21: 205-14
99. Hold G, Rabkin CS, Chow WH, Smith MG, Gammon MD, Sisch HA, et al. A gunctional polymorphisms of Toll like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 2007; 132: 905-12.
100. Hisida A, Matsuo K, Goto Y, Mitsuda Y, Hiraki K, Naito M, et al. Toll-like Receptor 4+3725G/C polymorphisms, Helicobacter pylori seropositivity and the risk of gastric atrophy and gastric cancer. Helicobacter 2009; 14: 47-53.
101. D´Elios MM, Amedi A, Benagiano M, Azzurri A, Del Prete G. Helicobacter pylori, T cells and cytokines: the "dangerous liaisons". FEMS Immunol Med Microbiol 2005; 44: 113-9.
102. Lehmann FS, Terracciano L, Carena I, Baeriswyl C, Drewe J, Tornillo L, et al. In situ correlation of cytokine secretion and apoptosis in Helicobacter pylori-associated gastritis. Am J Physiol Gastrointest Liver Physiol 2002; 283: G481-G488.
103. Holcombe C. Helicobacter pylori: The African enigma. Gut 1992; 33: 429-31.
104. Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, et al. The prevalence of Th17 cells in patients with gastric cáncer. Bioch Bioph Res Com 2008; 374: 533-7.
105. Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of Cag A phosphorilation for clinical aislates of Helicobacter pylori. Gastroenterology 2004; 127: 514-23.
106. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo C, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 95-200.
107. Wu MS, Chen CJ, Lin JT. Host environment interactions: their impact on progression from gastric inflammation to carcinogenesis and the development to new approaches to prevent and treat gastric cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 1878-82.
108. Amieva MR, El-Omar EM. Host bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008; 134: 306-23.
109. Ferreira AC, Isomoto H, Moriyama M, Fujioka T, Machado JC, Yamaoka Y. Helicobacter and gastric malignancies. Helicobacter 2008; 13 (Suppl 1): 28-34.
110. Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321-33.
111. Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol 1991; 261: G559-64.
112. El-Omar EM, Carrington M, Chow WH, McColl KEL, Bream JH, Young HA, et al. Interleukin-1 plymorphims associated with increased risk of gastric cancer. Nature 2000; 404: 398-402.
113. Machado JC, Pharoah P, Sousa S, Carvalho R, Oliveira C, Figuereido C, et al. Interleukin-1b and interleukin-1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001; 121: 823-9.
114. Chen A, Li CN, Hsu PI, Lai KH, Tseng HH, Hsu PN, et al. Risks of interleukin-1 genetic polymorphisms and Helicobacter pylori infection in the development of gastric cancer. Aliment Pharmacol Ther 2004; 20: 203-11.
115. Lahner E, Corleto VD, D´Ambra G, Di Giulio E, D´Elle Fave G, Anibale B. Is Interleukin 1 genotyping useful for the clinical management of patients with atrophic body gastritis? Aliment Phrmacol ther 2008; 27: 355-65.
116. Palli D, Saieva C, Luzzi I, Masala G, Topa S, Sera F, et al. Interleukin- 1 gene polymorphisms and gastric cancer risk in a high-risk Italian population. Am J Gastroenterol 2005; 100: 1941-8.
117. Lee SG, Kim B, Choi W, Lee I, Choi J, Song K. Lack of association between pro-inflammatory genotypes of the interleukin-1 (IL-1B -31 C/+ and IL-1RN *2/*2) and gastric cancer/duodenal ulcer in Korean population. Cytokine 2003; 21: 167-71.
118. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: a meta-analysis. Int J cancer 2007; 120: 552-62.
119. Camargo MC, Mera R, Correa P, Peek Jr RM, Fontham ET, Goodman KJ, et al. Interlukin-1beta and antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1674-87.
120. Kamangar F, Cheng C, Abnet CC, Rabkin C. Interleukin -1B polymorphisms and gastric cancer risk-A Metanalysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1920-8.
121. Meining A, Morgner A, Miehlke S, Morgner A, Bayerdorffer E, Stolte M. Atrophy-metaplasia-dysplasia-carcinoma sequence in the stomach: a reality or merely a hypothesis? Best Pract Res Gastroenterol 2001; 15: 983-98.
122. Filipe MI, Muñoz N, Matko I, Kato I, Pompe-Kim V, Jutersek A, et al. Intestinal metaplasia types and the risk of gastric cancer: A cohort in Slovenia. Int J Cancer 1994; 57: 324-9.
123. Ectors N, Dixon MF. The pronostic value of sulphomucin positive intestinal metaplasia in the deveopment of gastric cancer. Histopahology 1986; 10: 1271-7.
124. El Zimaity HM, Ramchatesingh J, Saeed MA, Hraham DY. Gastric intestinal metaplasia: subtypes and natural history. J Clin Pathol 2001; 54: 679-83.
125. Kakinoki R, Kushima R, Matsubara A, Saito Y, Okabe H, Fujiyama Y et al. Re-evaluation of histogenesis of gastric carcinoma: A comparative histopathological study between Helicobacter pylori-negative and H. pylori-positive cases. Dig Dis Sci 2009; 54: 614-20.
126. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrowderived cells. Science 2004; 306: 1568-71.
127. Li HC, Stoicov C, Rogers AB, Houghton J. Stem cells and cancer: evidence for bone marrow stem cells in epithelial cancers. World J Gastroenterol 2006; 12: 363-71.
128. Houghton J, Morozov A, Smirnova I, Wang TC. Stem Cell and Cancer. Seminars Cancer Biol 2007; 17: 191-203.
129. Takaishi S, Okumura T, Wang TC. Gastric cancer Stem Cell. J Clin Oncol 2008; 26: 2876-82.
130. Azuma T. Role of Cag A in the Helicobacter pylori infection and pathology. En Wang TC, (Eds). The biology of gastric cancers. Springer Science+Business Media, LLC 2009. p. 389-401.
131. Harcker J, Kaper JB. Pathogenicity Islands and the Evolution of Microbes. Ann Rev Microbiol 2000; 54: 641-679.
132. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. Cag A pathogenicity islad of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U.S.A 1996; 93: 14648-53.
133. Crowe SE. Helicobacter pylori infection, chronic inflammation and the development of malignancy. Curr Op Gastroenterol 2005; 21: 32-38.
134. Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 1573-81.
135. Handa O, Naito Y, Yoshikawa T. CagA protein of Helicobacter pylori: A hijacker of gastric epithelial cell signalin. Bioch Pharmacol 2007; 73: 1697-1702.
136. Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA Phosphorylation-Independent function in epithelial proliferation and inflammation. Cell Host Micr 2009; 5: 23-34.
137. Steffen Backert and Matthias Selbach. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 8: 1573-1581.
138. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4: 688-94.
139. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natnl Acad Sci 2008; 105: 1003-8.
140. Yamaoka Y, Kato M, Asaka M. Geographic differences in gastric cancer can be explained by differences between Helicobacter pylori Straits. Intern Med 2008; 47: 1077-83.
141. Fox JG, Wang TC. Inflammation atrophy and gastric cancer. J Clin Invest 2007; 17: 60-9.
142. Cover TL, Tummuru MKR, Cao P, Thompson SA, Blaser MJ. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori Straits. J Biol Chem 1994; 269: 1566-573.
143. Yuan JP, Li T, Chen HB, Li ZH, Yang GZ, Hu BH, et al. Analysis of gene expression profile in gastric cancer cells stimulated with Helicobacter pylori isogenic strains. J Med Microbiol 2004; 53: 965-74.
144. Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori peptic ulceration. J Biol Chem 1995; 270: 1771-7.
145. Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infet 2009 Early Rel (acceso junio 10, 2009).
146. Letley DP, Atherton JC. Natural Diversity in the n terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J Bacteriol 2000; 182: 3278-80.
147. Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh M, et al. A new Helicobacter pylori Vacuolating cytotoxin determinant the intermediate region is associated with gastric cancer. Gastroenterology 2007; 133: 926-36.
148. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesion binding fucosylated histo-blood group antigens revealed by retagging. Science 1998; 279: 373-7.
149. Peek Jr RM, Thompson SA, Donahue JP, Tham KT, Athertton JC, Blaser MJ, et al. Adherence to gastric epithelial cells induces expression of a Helicobacter pylori gene ice A, that is associated with clinical outcome. Proc Assoc Am Phys 1998; 110: 531-44.
150. Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsiveelement-like element in inteleukin8 promoter in Helicobacter pylori infection. Gastroenterology 2004; 126: 1030-45.
151. Yamaoka Y, Kwon DH, Graham DY, A M (r). 34 000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA 2000; 97: 7533-8.
152. Kontouras J, Zavos C, Chatzopoulos D, Katsinelos P. New aspects of Helicobacter pylori infection involvement in gastric oncogenesis. J Surg Res 2008; 146: 149-58.
153. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003; 124: 1193-1201.
154. Beales IL, Calam J. Interleukin 1 beta and tumor necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998; 42: 227-34.
155. Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor (alpha) and risk of gastric cancer in a Chinese population. Carcinogenesis 2005; 26: 631-6.
156. Ohyauchi M, Imatani A, Yonechi M, Asano N, Miura A, Lijima K, et al. The polymorphisms inerleukin 8-251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut 2005; 54: 330-5.
157. Malfertheiner P, Megraud F, O´Morain C Bazzoli F, El-Omar E, Graham DY, et al. Current concepts in the management of Helicobacter pylori infection- The Maastricht III consensus report. Gut 2007; 56: 772-81.
158. Wong BC, Lam FK, Wong WM, Chen JS, Zheng TT, Fen RE, et al. Helicobacter pylori eradication to prevent gastric cancer in high risk region in China: a randomized controlled trial. JAMA 2004; 291: 187-94.
159. Lochhead P, El-Omar EM. Helicobacter pylori infection and gastric cancer. Best Pract Res Clin Gastroenterol 2007; 21: 281-97.
160. De Vries AC, Kuipers EJ, Raws EAJ. Helicobacter pylori eradication and gastric cancer: when is the horse out of the barn? Am J Gastroenterol 2009; 104: 1342-5.
161. Zullo A, Hassan C, Andriani A, Cristoffari F, De Francesco V, Ierardi E, et al. Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: A pooled Data Analysis. Am J Gastroenterol 2009 advance online publication 16 june 2009.
162. Malferthteiner P, Schulktze V, Rosenkranz B, Kauffman SHE, Ulrichz T, Novicki D, et al. Safety and inmunogenecity of an intramuscular Helicobacter pylori vaccine in noninfected volunteers: a phase I study. Gastroenterology 2008; 135: 787-95.
1. Ferlay J, Bray F, Pisani P, et al. GLOBOCAN 2002: Cancer Incidence, Mortality and Prevalence Worldwide, versión 2.0 IARC CancerBase No 5 Lyon: IARC 2004. [ Links ]
2. Parkin DM, Bray F, Ferlay J, Paisani P. Global Cancer Statistics 2005; CA Cancer J Clin 2005; 55: 74-108. [ Links ]
3. Krew KD, Neugut AI. Epidemiology of gastric cancer. World J Gastroenterol 2006; 12: 354-62. [ Links ]
4. Hamashima C, Shibuya D, Yamazaki H, Inoue K, Fukao A, Saito Sobue T. The Japanese guidelines for gastric cancer screening. Jpn J Clin Oncol 2008; 38: 259-67. [ Links ]
5. Piñeros M, Hernández G, Bray F. Increasing mortality rates of common malignancies in Colombia. Cancer 2004; 101: 2285-92. [ Links ]
6. Correa P, Piazzuelo MB, Camargo MC. Overview and pathology of gastric cancer. En Wang T, Fox J, Giraud A. (Edit). The biology of gastric cancers. Springer Science Business + Media 2009. p. 21-44. [ Links ]
7. Smith MG, Hold GL, Tahara E, El-Omar EM. Cellular and molecular aspects of gastric cancer. World J Gastroenterol 2006; 12: 2979-90. [ Links ]
8. Mosss SF, Malfertheiner P. Helicobacter and gastric malignancies. Helicobacter 2007; 12(Suppl 1): 23-30. [ Links ]
9. Farinha P, Gascoyne RD. Helicobacter pylori and MALT lymphoma. Gastroenterology 2005; 128: 1579-1605. [ Links ]
10. Jarvi O, Lauren P. On the role of heterotopias of the intestinal epithelium in the pathogenesis of gastric cancer. Acta Pathol Microbiol Scand 1951; 29: 26-44. [ Links ]
11. Lauren, P. The two histological main types of gastric carcinoma: diffuse and so-called intestinal-type carcinoma. An attempt at a histo-clinical classification. Acta Pathol Microbiol Scand 1965; 64: 31-49. [ Links ]
12. Correa P, Carneiro F. Classification of gastric carcinomas. Curr Diagn Pathol 1997; 4: 51-9. [ Links ]
13. Ekstrom AM, Serafini M, Nyren O, Hansson LE, Ye W, Wolk A. Dietary antioxidante intake and the risk of noncardia cancer for the intestinal and diffuse types: a population-based case-control study in Sweden. Int J Cancer 2000; 87: 133-40. [ Links ]
14. El-Omar EM, Lochhead P. Gastric cancer. Br Med Bull 2008; 85: 87-100. [ Links ]
15. Henson DE, Dittus C, Younes M, Nagyen H, Albores-Saavedra J. Differential trends in the intestinal and diffuse topless of gastric carcinoma in the United States-2000: increase in the signet ring cell type. Arch Pathol Lab Med 2004; 128: 765-70. [ Links ]
16. Oliveira C, Seruca R, Carneiro F. Hereditary gastric cancer. Best Pract Res Clin Gastroenterol 2009; 23: 147-57. [ Links ]
17. Alberts SR, Cervantes A, van de Velde CJ. Gastric cancer: epidemiology, pathology and treatment. Ann Oncol 2003; 14 (suppl 2): ii31-ii36. [ Links ]
18. Guilford P, Hopkins J, Harraway J, McLeod M, McLeod N, Harawira P, Taite H, Scoular R, Miller A, Reeve AE. E-cadherin germline mutations in familial gastric cancer. Nature 1998; 392: 402-5. [ Links ]
19. Carneiro F, Sobrinho-Simoes M. Hereditary diffuse gastric cancer: lessons from histopatholgy. Adv Anat Pathol 2005; 12: 151-2. [ Links ]
20. Medina-Franco H, Barreto-Zúñiga R, García-Álvarez MN. Preemptive total gastrectomy for hereditary gastric cancer. J Gastrointest Surg 2007; 11: 314-7. [ Links ]
21. Medina-Franco H, Medina AR, Vizcaíno G, Medina-Franco JL. Single nucleotide polymorphisms in the promoter region of the E-cadherin gene in gastric cancer: case-control study in a young Mexican population. Ann Surg Oncol 2007; 14: 2246-9. [ Links ]
22. Cisco RM, Ford JM, Norton JA. Hereditary diffuse gastric cancer. Cancer 2008; 113 (7 Suppl): 1850-6. [ Links ]
23. Masciari S, Larsson N, Senz J, Boid N, Kaurah P, Kandel MJ, et al. Germline E-cadherin mutations in familial lobular breast cancer. J Med Genet 2007; 44: 726-31. [ Links ]
24. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg Pathol 1995; 19 (Suppl.1): S37-S43. [ Links ]
25. Kabir S. Effect of Helicobacter pylori eradication on incident of gastric cancer in human and animal models: underlying biochemical and molecular events. Helicobacter 2009; 14: 159-71. [ Links ]
26. Correa P, Chen VW. Gastric Cancer. Cancer Surv 1994; 19: 55-76. [ Links ]
27. Kono S, Irohata T. Nutrition and stomach cancer. Cancer Causes Control 1996; 7: 41-55. [ Links ]
28. Larsson SC, Bergkvist L, Wolk A. Fruit and vegetable consumption and incidence of gastric cancer: A prospective study. Cancer Epidemiol Biomarkers Prev 2006; 15: 1998-2001. [ Links ]
29. Bae JM, Lee EJ, Guyatt G. Citrus fruit intake and stomach cancer risk: a quantitative systematic review. Gastric Cancer 2008; 11: 23-37. [ Links ]
30. González CA, Pera G, Agudo A, Bueno-de Mezquita HB, Ceroti M, et al. Fruit and vegetable intake and the risk of stomach and oesophagus adenocarcinoma in the European Prospective Investigation into Cancer and Nutrition (EPIC-Eurogast). Int J Cancer 2006; 118: 2559-66. [ Links ]
31. Bjelakovic G, Nikolova D, Simonetti RG, Gluud C. Antioxidant supplements for preventing gastrointestinal cancers. Cochrane Database of Systematic Reviews 2004; Issue 4. [ Links ]
32. Liu C, Huang XD, Russell RM. Diet and gastric cancer. En Wang TC, Fox J, Giraud A. (Edits). The biology of gastric cancers. Springer Science+Business Media LLC 2009. p. 59-89. [ Links ]
33. Correa P, Fontham ET, Bravo JC, Bravo LE, Ruiz B, Zarama G, Realpe JL, Malcom GT, Li D, Johnson WD, Mera R. Chemoprevention of gastric dysplasia: randomized trial of antioxidant supplements and anti-helicobacter pylori therapy. J Natl Cancer Inst 2000; 92: 1881-8. [ Links ]
34. Mera R, Fontham ET, Bravo LE, Bravo JC, Piazuelo MB, Camargo MC, et al. Long term follow up of patients treated for Helicobacter pylori infection. Gut. 2005; 54: 1536-40. [ Links ]
35. Blot WJ, Li JY, Taylor PR, Guo W, Dawsey S, Wang GQ, Yang CS, Zheng SF, Gail M, Li GY, et al. Nutrition intervention trials in Linxian, China: supplementation with specific vitamin/mineral combinations, cancer incidence, and disease-specific mortality in the general population. J Natl Cancer Inst 1993; 85: 1483-92. [ Links ]
36. Goswami UC, Sharma N. Efficiency of a few retinoids and carotenoids in vivo in controlling benzo[a]pyrene-induced forestomach tumour in female Swiss mice. Br J Nutr 2005; 94: 540-3. [ Links ]
37. Velmurugan B, Bhuvaneswari V, Burra UK, Nagini S. Prevention of N-methyl-N′-nitro-N-nitrosoguanidine and saturated sodium chloride-induced gastric carcinogenesis in Wistar rats by lycopene. Eur J Cancer Prev 2002; 11: 19-26. [ Links ]
38. Velmurugan B, Bhuvaneswari V, Nagini S. Antiperoxidative effects of lycopene during N-methyl-N′-nitro-N-nitrosoguanidine-induced gastric carcinogenesis. Fitoterapia 2002; 73: 604-11. [ Links ]
39. Velmurugan B, Mani A, Nagini S. Combination of S-allylcysteine and lycopene induces apoptosis by modulating Bcl-2, Bax, Bim and caspases during experimental gastric carcinogenesis. Eur J Cancer Prev 2005; 14: 387-93. [ Links ]
40. Liu C, Russell RM, Wang XD. Lycopene supplementation prevents smoke-induced changes in p53, p53 phosphorylation, cell proliferation, and apoptosis in the gastric mucosa of ferrets. J Nutr 2006; 136: 106-11. [ Links ]
41. Wang X, Willen R, Wadstrom T. Astaxanthin-rich algal meal and vitamin C inhibit Helicobacter pylori infection in BALB/cA mice. Antimicrob Agents Chemother 2000; 44: 2452-7. [ Links ]
42. Liu BH, Lee YK. Effect of total secondary carotenoids extracts from Chlorococcum sp on Helicobacter pylori-infected BALB/c mice. Int Immunopharmacol 2003; 3: 979-86. [ Links ]
43. World Cancer Research Fund, American Institute for Cancer Research. Food, nutrition and the prevention of cancer: a global perspective. Washington, DC: American Institute for cancer Research 1997. [ Links ]
44. Kato S, Tsukamoto T, Mizoshita T, Tanaka H, Kumagai T, Ota H, Katsuyama T, Asaka M, Tatematsu M. High salt diets dose-dependently promote gastric chemical carcinogenesis in Helicobacter pylori-infected Mongolian gerbils associated with a shift in mucin production from glandular to surface mucous cells. Int J Cancer 2006; 119: 1558-66. [ Links ]
45. Watanabe H, Takahashi T, Okamoto T, Ogundigie PO, Ito A. Effects of sodium chloride and ethanol on stomach tumorigenesis in ACI rats treated with N-methyl-N′-nitro-Nnitrosoguanidine: a quantitative morphometric approach. Jpn J Cancer Res 1992; 83: 588-93. [ Links ]
46. Furihata C, Sato Y, Hosaka M, Matsushima T, Furukawa F, Takahashi M. NaCl induced ornithine decarboxylase and DNA synthesis in rat stomach mucosa. Biochem Biophys Res Commun 1984; 121: 1027-32. [ Links ]
47. Sorbye H, Kvinnsland S, Svanes K. Effect of salt-induced mucosal damage and healing on penetration of N-methyl-N′-nitro-N-nitrosoguanidine to proliferative cells in the gastric mucosa of rats. Carcinogenesis 1994; 15: 673-9. [ Links ]
48. Fox JG, Dangler CA, Taylor NS, King A, Koh TJ, Wang TC. High-salt diet induces gastric epithelial hyperplasia and parietal cell loss, and enhances Helicobacter pylori colonization in C57BL/6 mice. Cancer Res 1999; 59: 4823-8. [ Links ]
49. Jakszyn P, González, CA. Nitrosamine and related food intake and gastric and oesophageal cancer risk: a systematic review of the epidemiological evidence. World J Gastroenterol 2006; 12: 4296-303. [ Links ]
50. Mirvish SS. Role of N-nitroso compounds (NOC) and N-nitrosation in etiology of gastric, esophageal, nasopharyngeal and bladder cancer and contribution to cancer of known exposures to NOC. Cancer Lett.1995; 93: 17-48. [ Links ]
51. Jakszyn P, Bingham S, Pera G, Agudo A, Luben R, Welch A, et al. Endogenous versus exogenous exposure to N-nitroso compounds and gastric cancer risk in the European Prospective Investigation into Cancer and Nutrition (EPIC-EURGAST) study. Carcinogenesis.2006; 27: 1497-501. [ Links ]
52. Schistosomes, Liver fluyes and Helicobacter pylori IARC Working group on the evaluation of carcinogenic risks to humans. Lyon, 7-14 June 1994. IARC Monog Eval Carcinog Risks Hum 1994; 61: 1-41. [ Links ]
53. Forman D, Newell DG, Fullerton F, Yarnell JW, Stacey AR, Wald N, et al. Association between infection with Helicobacter pylori and risk of gastric cancer: evidence from a prospective investigation. BMJ 1991, 302: 1302-5. [ Links ]
54. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, Orentrich N, et al. Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 1991; 325: 1127-31. [ Links ]
55. Nomura A, Stemmermann GN, Chyou PH, Kato I, Perez-Perez GI, Blaser M, et al. Helicobacter pylori infection and gastric carcinoma among Japanese Americans in Hawaii. N Engl J Med 1991; 325: 1132-6. [ Links ]
56. Sugiyama A, Maruta F, Ikeno T, Ishida K, Kawasaki S, Katsuyama T, et al. Helicobacter pylori infection enhances N-methyl-N-nitrosourea-induced stomach carcinogenesis in the Mongolian gerbil. Cancer Res 1998; 58: 2067-69. [ Links ]
57. Watanabe T, Tada M, Nagai H, Sasaki S, Nakao M. Helicobacter pylori infection induces gastric cancer in Mongolian gerbils. Gastroenterology 1998; 115: 642-8. [ Links ]
58. An International association between Helicobacter pylori infection and gastric cancer. The EUROGAST Study Group. Lancet 1993; 341: 1359-62. [ Links ]
59. Huang JQ, Zheng GF, Sumanak K, Irvine EJ, Hunt RH. Meta-analysis of the relationship between cag A seropositivity and gastric cancer. Gastroenterology 2003; 125: 1636-44. [ Links ]
60. Peter S, Begglinnger C. Helicobacter pylori and gastric cancer: The causal relationship. Digestion 2007; 75: 25-35. [ Links ]
61. Suerbaum S, Michetti P. Helicobacter pylori infection. N Engl J Med 2002; 347: 1175-86. [ Links ]
62. Mueller A, Falkow S, Amieva MR. Helicobacter pylori and gastric cancer: what can be learned by studying the response of gastric epithelial cells to the infection? Cancer Epidemiol Biomarkers Prev 2005; 14: 1859-64. [ Links ]
63. Peter S, Beglinger C. Helicobacter pylori and gastric cancer: the causal relationship. Digestion 2007; 75: 25-35. [ Links ]
64. Wu MS, Shun CT, Wu CC, et al. Epstein Barr virus associated with Helicobacter pylori. Infection and genetic alterations. Gastroenterology 2000; 118: 1031-8. [ Links ]
65. Report of the Digestive Health Initiative International Update. Conference on Helicobacter pylori. Gastroenterology 1997; 113 (Suppl): S4-S8. [ Links ]
66. Saad R, Chey W. A clinicians guide to managing Helicobacter pylori infection. Clev Clin J Med 2005; 72: 109-124. [ Links ]
67. Parsonnet J. Helicobacter pylori: the size of the problem. Gut 1998; 43: S6-S9. [ Links ]
68. Correa P, Schneider BG. Etiology of gastric cancer: what is new? Cancer Epidemiol Biomarkers Prev 2005; 14: 1865-8. [ Links ]
69. Guillen D, McColl KEL. Gastroduodenal disease, Helicobacter pylori, and genetic polymorphisms. Clinical Gastroenterol Hepatol 2005; 3: 1180-86. [ Links ]
70. El Omar EM, Penman I, Ardill JE, Chittajallu RS, Howie C, Mc Coll KE, et al. Helicobacter pylori infection and abnormalities of acid secretion in patients with duodenal ulcer disease. Gastroenterology 1995; 109:681-91. [ Links ]
71. Lai LH, Sung JJY. Helicobacter pylori and benign upper digestive disease. Best Pract Res Clin Gastroenterol 2007; 21: 261-79. [ Links ]
72. Faber K. Chronic gastritis: its relation to achylia and ulcer. Lancet 1927; 2: 902-7. [ Links ]
73. Graham DY, Lu H, Yamaoka Y. African, Asian or Indian enigma, the East Asian Helicobacter pylori: facts or medical myths. J Dig Dis 2009; 10: 77-84. [ Links ]
74. Uemura N, Okamoto S, Yamamoto S, Matsumura N, Yamaguchi S, Yamakido M, et al. Helicobacter pylori infection and the development of gastric cancer. N Engl J Med 2001; 345: 784-9. [ Links ]
75. Chiba T, Seno H, Marusawa Y, Okazaki K. Host factors are important in determining clinical outcomes of Helicobacter pylori infection. J Gastroenterol 2006; 41: 1-9. [ Links ]
76. Chiba T, Marusawa H, Seno H, Watanabe N. Mechanism Fort he gastric cancer development by Helicobacter infection. J Gastroenterol Hepatol 2008; 23: 1175-81. [ Links ]
77. Bonnevie O. The incidence of duodenal ulcer in Copenhagen County. Gastroenterology 1975; 10: 385-93. [ Links ]
78. Hu PJ, Li YY, Zhou MH, Chen MH, Huang BJ, Mitchel HM, et al. Helicobacter pylori associated with a high prevalence of duodenal ulcer disease and low prevalence of gastric cancer in a developing nation. Gut 1995; 36: 198-202. [ Links ]
79. Correa P. Helicobacter pylori and gastric carcinogenesis. Am J Surg pathol 1995; 19: S37-S43. [ Links ]
80. Ernst PB, Peura DA, Crowe SE. The translation of Helicobacter pylori Basic research to patient care. Gastroenterology 2006; 130: 188-206. [ Links ]
81. Matsukura N, Suzuki K, Kavachi T, Aoyagi M, Sugimura T, Kitaoka H, et al. Distribution of marker enzymes and mucin in intestinal metaplasia in human stomach and relation to complete and incomplete types of intestinal metaplasia to minute gastric carcinomas. J Natl Cancer Inst 1980; 65: 231-40. [ Links ]
82. Correa P, Hoghton J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007; 133: 659-72. [ Links ]
83. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: a morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 195-200. [ Links ]
84. Matsumoto Y, Marusawa H, Kinoshita K, Endo Y, Kou T. Helicobacter pylori infection triggers aberrant expresión of activation-induced cytidine deaminase in gastric epithelium. Nat Med 2007; 13: 470-6. [ Links ]
85. Takaishi S, Wang TC. Providing AID to p53 mutagenesis. Nat Med 2007; 13: 404-6. [ Links ]
86. Correa P, Haenszel W, Cuello C, Tannembaum S, Archer M. A model for gastric cancer epidemiology. Lancet 1975; 2: 58-60. [ Links ]
87. Warren JR, Marshall BJ. Unidentified curved bacilli on gastric epithelium in active chronic gastritis. Lancet 1983; 1: 1273-5. [ Links ]
88. Correa P. The epidemiology and pathogenesis of chronic: three etiologic entities. Front Gastrointest Res 1980; 6: 98-108. [ Links ]
89. Correa P. Clinical implications of recent development in gastric cancer pathology and epidemiology. Sem Oncol 1985; 12: 2-10. [ Links ]
90. Bonne C, Hartz PH, Klerks JV, Posthuma JH, Radsma W, Tjokronegoro S, et al. Morphology of the stomach and gastric secretion in Malays and Chinese and the different incident of gastric ulcer and cancer in these races. Am J Cancer 1938; 33: 265-79. [ Links ]
91. Siurala M, Varis K, Wiljasalo M. Studies of patients with atrophic gastritis: a 10 -15 year follow-up. Scand J Gastroenterol 1966; 1: 40-8. [ Links ]
92. Volk J, Parsonnet J. Epidemiology of Gastric Cancer. En Wang TC, Fox J, Giraud A. (Eds). The biology of Gastric cancers. Springer Science+ Business Media LLC 2009. p. 25-57. [ Links ]
93. Matysiak-Budnik T, Mégraud F. Helicobacter pylori infection and gastric cancer. Eur J Cancer 2006; 42: 708-16. [ Links ]
94. Girardin SE, Boneca IG, Carneiro LA, Antignac A, Jehanno M, Viala J, et al. NodI detect a unique muropeptide from gram negative bacterial peptidoglycan. Science 2003; 300: 1584-87. [ Links ]
95. Girardin SE Travassos LH, Herve M, Blanot D, Boneca IG, Philpott DJ, et al. Peptiglycan molecular requeriments allowing detection by NodI and Nod2. J Biol Chem 2003; 278: 41702-8. [ Links ]
96. Keeffe J, Moran AP. Conventional, regulatory and unconventional T cells in the immunologic response to Helicobacter pylori. Helicobacter 2008; 13: 1-19. [ Links ]
97. Ma J, Chen T, Mandelin J, Ceponis A, Miller NE. Regulation of macrophage activation. Cell Mol Life Sci 2003; 60: 2334-46. [ Links ]
98. Malaty HM. Epidemiology of Helicobacter pylori. Best Pract Res Clin Gastroenterol 2007; 21: 205-14. [ Links ]
99. Hold G, Rabkin CS, Chow WH, Smith MG, Gammon MD, Sisch HA, et al. A gunctional polymorphisms of Toll like receptor 4 gene increases risk of gastric carcinoma and its precursors. Gastroenterology 2007; 132: 905-12. [ Links ]
100. Hisida A, Matsuo K, Goto Y, Mitsuda Y, Hiraki K, Naito M, et al. Toll-like Receptor 4+3725G/C polymorphisms, Helicobacter pylori seropositivity and the risk of gastric atrophy and gastric cancer. Helicobacter 2009; 14: 47-53. [ Links ]
101. D´Elios MM, Amedi A, Benagiano M, Azzurri A, Del Prete G. Helicobacter pylori, T cells and cytokines: the "dangerous liaisons". FEMS Immunol Med Microbiol 2005; 44: 113-9. [ Links ]
102. Lehmann FS, Terracciano L, Carena I, Baeriswyl C, Drewe J, Tornillo L, et al. In situ correlation of cytokine secretion and apoptosis in Helicobacter pylori-associated gastritis. Am J Physiol Gastrointest Liver Physiol 2002; 283: G481-G488. [ Links ]
103. Holcombe C. Helicobacter pylori: The African enigma. Gut 1992; 33: 429-31. [ Links ]
104. Zhang B, Rong G, Wei H, Zhang M, Bi J, Ma L, et al. The prevalence of Th17 cells in patients with gastric cáncer. Bioch Bioph Res Com 2008; 374: 533-7. [ Links ]
105. Argent RH, Kidd M, Owen RJ, Thomas RJ, Limb MC, Atherton JC. Determinants and consequences of different levels of Cag A phosphorilation for clinical aislates of Helicobacter pylori. Gastroenterology 2004; 127: 514-23. [ Links ]
106. Farinati F, Cardin R, Cassaro M, Bortolami M, Nitti D, Tieppo C, et al. Helicobacter pylori, inflammation, oxidative damage and gastric cancer: morphological, biological and molecular pathway. Eur J Cancer Prev 2008; 17: 95-200. [ Links ]
107. Wu MS, Chen CJ, Lin JT. Host environment interactions: their impact on progression from gastric inflammation to carcinogenesis and the development to new approaches to prevent and treat gastric cancer. Cancer Epidemiol Biomarkers Prev 2005; 14: 1878-82. [ Links ]
108. Amieva MR, El-Omar EM. Host bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008; 134: 306-23. [ Links ]
109. Ferreira AC, Isomoto H, Moriyama M, Fujioka T, Machado JC, Yamaoka Y. Helicobacter and gastric malignancies. Helicobacter 2008; 13 (Suppl 1): 28-34. [ Links ]
110. Blaser MJ, Atherton JC. Helicobacter pylori persistence: biology and disease. J Clin Invest 2004; 113: 321-33. [ Links ]
111. Wallace JL, Cucala M, Mugridge K, Parente L. Secretagogue-specific effects of interleukin-1 on gastric acid secretion. Am J Physiol 1991; 261: G559-64. [ Links ]
112. El-Omar EM, Carrington M, Chow WH, McColl KEL, Bream JH, Young HA, et al. Interleukin-1 plymorphims associated with increased risk of gastric cancer. Nature 2000; 404: 398-402. [ Links ]
113. Machado JC, Pharoah P, Sousa S, Carvalho R, Oliveira C, Figuereido C, et al. Interleukin-1b and interleukin-1RN polymorphisms are associated with increased risk of gastric carcinoma. Gastroenterology 2001; 121: 823-9. [ Links ]
114. Chen A, Li CN, Hsu PI, Lai KH, Tseng HH, Hsu PN, et al. Risks of interleukin-1 genetic polymorphisms and Helicobacter pylori infection in the development of gastric cancer. Aliment Pharmacol Ther 2004; 20: 203-11. [ Links ]
115. Lahner E, Corleto VD, D´Ambra G, Di Giulio E, D´Elle Fave G, Anibale B. Is Interleukin 1 genotyping useful for the clinical management of patients with atrophic body gastritis? Aliment Phrmacol ther 2008; 27: 355-65. [ Links ]
116. Palli D, Saieva C, Luzzi I, Masala G, Topa S, Sera F, et al. Interleukin- 1 gene polymorphisms and gastric cancer risk in a high-risk Italian population. Am J Gastroenterol 2005; 100: 1941-8. [ Links ]
117. Lee SG, Kim B, Choi W, Lee I, Choi J, Song K. Lack of association between pro-inflammatory genotypes of the interleukin-1 (IL-1B -31 C/+ and IL-1RN *2/*2) and gastric cancer/duodenal ulcer in Korean population. Cytokine 2003; 21: 167-71. [ Links ]
118. Wang P, Xia HH, Zhang JY, Dai LP, Xu XQ, Wang KJ. Association of interleukin-1 gene polymorphisms with gastric cancer: a meta-analysis. Int J cancer 2007; 120: 552-62. [ Links ]
119. Camargo MC, Mera R, Correa P, Peek Jr RM, Fontham ET, Goodman KJ, et al. Interlukin-1beta and antagonist gene polymorphisms and gastric cancer: a meta-analysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1674-87. [ Links ]
120. Kamangar F, Cheng C, Abnet CC, Rabkin C. Interleukin -1B polymorphisms and gastric cancer risk-A Metanalysis. Cancer Epidemiol Biomarkers Prev 2006; 15: 1920-8. [ Links ]
121. Meining A, Morgner A, Miehlke S, Morgner A, Bayerdorffer E, Stolte M. Atrophy-metaplasia-dysplasia-carcinoma sequence in the stomach: a reality or merely a hypothesis? Best Pract Res Gastroenterol 2001; 15: 983-98. [ Links ]
122. Filipe MI, Muñoz N, Matko I, Kato I, Pompe-Kim V, Jutersek A, et al. Intestinal metaplasia types and the risk of gastric cancer: A cohort in Slovenia. Int J Cancer 1994; 57: 324-9. [ Links ]
123. Ectors N, Dixon MF. The pronostic value of sulphomucin positive intestinal metaplasia in the deveopment of gastric cancer. Histopahology 1986; 10: 1271-7. [ Links ]
124. El Zimaity HM, Ramchatesingh J, Saeed MA, Hraham DY. Gastric intestinal metaplasia: subtypes and natural history. J Clin Pathol 2001; 54: 679-83. [ Links ]
125. Kakinoki R, Kushima R, Matsubara A, Saito Y, Okabe H, Fujiyama Y et al. Re-evaluation of histogenesis of gastric carcinoma: A comparative histopathological study between Helicobacter pylori-negative and H. pylori-positive cases. Dig Dis Sci 2009; 54: 614-20. [ Links ]
126. Houghton J, Stoicov C, Nomura S, Rogers AB, Carlson J, Li H, Cai X, Fox JG, Goldenring JR, Wang TC. Gastric cancer originating from bone marrowderived cells. Science 2004; 306: 1568-71. [ Links ]
127. Li HC, Stoicov C, Rogers AB, Houghton J. Stem cells and cancer: evidence for bone marrow stem cells in epithelial cancers. World J Gastroenterol 2006; 12: 363-71. [ Links ]
128. Houghton J, Morozov A, Smirnova I, Wang TC. Stem Cell and Cancer. Seminars Cancer Biol 2007; 17: 191-203. [ Links ]
129. Takaishi S, Okumura T, Wang TC. Gastric cancer Stem Cell. J Clin Oncol 2008; 26: 2876-82. [ Links ]
130. Azuma T. Role of Cag A in the Helicobacter pylori infection and pathology. En Wang TC, (Eds). The biology of gastric cancers. Springer Science+Business Media, LLC 2009. p. 389-401. [ Links ]
131. Harcker J, Kaper JB. Pathogenicity Islands and the Evolution of Microbes. Ann Rev Microbiol 2000; 54: 641-679. [ Links ]
132. Censini S, Lange C, Xiang Z, Crabtree JE, Ghiara P, Borodovsky M, et al. Cag A pathogenicity islad of Helicobacter pylori, encodes type I-specific and disease-associated virulence factors. Proc Natl Acad Sci U.S.A 1996; 93: 14648-53. [ Links ]
133. Crowe SE. Helicobacter pylori infection, chronic inflammation and the development of malignancy. Curr Op Gastroenterol 2005; 21: 32-38. [ Links ]
134. Backert S, Selbach M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 1573-81. [ Links ]
135. Handa O, Naito Y, Yoshikawa T. CagA protein of Helicobacter pylori: A hijacker of gastric epithelial cell signalin. Bioch Pharmacol 2007; 73: 1697-1702. [ Links ]
136. Suzuki M, Mimuro H, Kiga K, Fukumatsu M, Ishijima N, Morikawa H, et al. Helicobacter pylori CagA Phosphorylation-Independent function in epithelial proliferation and inflammation. Cell Host Micr 2009; 5: 23-34. [ Links ]
137. Steffen Backert and Matthias Selbach. Role of type IV secretion in Helicobacter pylori pathogenesis. Cel Microbiol 2008; 10: 8: 1573-1581. [ Links ]
138. Hatakeyama M. Oncogenic mechanisms of the Helicobacter pylori CagA protein. Nat Rev Cancer 2004; 4: 688-94. [ Links ]
139. Ohnishi N, Yuasa H, Tanaka S, Sawa H, Miura M, Matsui A, et al. Transgenic expression of Helicobacter pylori CagA induces gastrointestinal and hematopoietic neoplasms in mouse. Proc Natnl Acad Sci 2008; 105: 1003-8. [ Links ]
140. Yamaoka Y, Kato M, Asaka M. Geographic differences in gastric cancer can be explained by differences between Helicobacter pylori Straits. Intern Med 2008; 47: 1077-83. [ Links ]
141. Fox JG, Wang TC. Inflammation atrophy and gastric cancer. J Clin Invest 2007; 17: 60-9. [ Links ]
142. Cover TL, Tummuru MKR, Cao P, Thompson SA, Blaser MJ. Divergence of genetic sequences for the vacuolating cytotoxin among Helicobacter pylori Straits. J Biol Chem 1994; 269: 1566-573. [ Links ]
143. Yuan JP, Li T, Chen HB, Li ZH, Yang GZ, Hu BH, et al. Analysis of gene expression profile in gastric cancer cells stimulated with Helicobacter pylori isogenic strains. J Med Microbiol 2004; 53: 965-74. [ Links ]
144. Atherton JC, Cao P, Peek RM Jr, Tummuru MK, Blaser MJ, Cover TL. Mosaicism in vacuolating cytotoxin alleles of Helicobacter pylori peptic ulceration. J Biol Chem 1995; 270: 1771-7. [ Links ]
145. Sugimoto M, Yamaoka Y. The association of vacA genotype and Helicobacter pylori-related disease in Latin American and African populations. Clin Microbiol Infet 2009 Early Rel (acceso junio 10, 2009). [ Links ]
146. Letley DP, Atherton JC. Natural Diversity in the n terminus of the mature vacuolating cytotoxin of Helicobacter pylori determines cytotoxin activity. J Bacteriol 2000; 182: 3278-80. [ Links ]
147. Rhead JL, Letley DP, Mohammadi M, Hussein N, Mohagheghi MA, Eshagh M, et al. A new Helicobacter pylori Vacuolating cytotoxin determinant the intermediate region is associated with gastric cancer. Gastroenterology 2007; 133: 926-36. [ Links ]
148. Ilver D, Arnqvist A, Ogren J, Frick IM, Kersulyte D, Incecik ET, et al. Helicobacter pylori adhesion binding fucosylated histo-blood group antigens revealed by retagging. Science 1998; 279: 373-7. [ Links ]
149. Peek Jr RM, Thompson SA, Donahue JP, Tham KT, Athertton JC, Blaser MJ, et al. Adherence to gastric epithelial cells induces expression of a Helicobacter pylori gene ice A, that is associated with clinical outcome. Proc Assoc Am Phys 1998; 110: 531-44. [ Links ]
150. Yamaoka Y, Kudo T, Lu H, Casola A, Brasier AR, Graham DY. Role of interferon-stimulated responsiveelement-like element in inteleukin8 promoter in Helicobacter pylori infection. Gastroenterology 2004; 126: 1030-45. [ Links ]
151. Yamaoka Y, Kwon DH, Graham DY, A M (r). 34 000 proinflammatory outer membrane protein (oipA) of Helicobacter pylori. Proc Natl Acad Sci USA 2000; 97: 7533-8. [ Links ]
152. Kontouras J, Zavos C, Chatzopoulos D, Katsinelos P. New aspects of Helicobacter pylori infection involvement in gastric oncogenesis. J Surg Res 2008; 146: 149-58. [ Links ]
153. El-Omar EM, Rabkin CS, Gammon MD, Vaughan TL, Risch HA, Schoenberg JB, et al. Increased risk of noncardia gastric cancer associated with proinflammatory cytokine gene polymorphisms. Gastroenterology 2003; 124: 1193-1201. [ Links ]
154. Beales IL, Calam J. Interleukin 1 beta and tumor necrosis factor alpha inhibit acid secretion in cultured rabbit parietal cells by multiple pathways. Gut 1998; 42: 227-34. [ Links ]
155. Lu W, Pan K, Zhang L, Lin D, Miao X, You W. Genetic polymorphisms of interleukin (IL)-1B, IL-1RN, IL-8, IL-10 and tumor necrosis factor (alpha) and risk of gastric cancer in a Chinese population. Carcinogenesis 2005; 26: 631-6. [ Links ]
156. Ohyauchi M, Imatani A, Yonechi M, Asano N, Miura A, Lijima K, et al. The polymorphisms inerleukin 8-251 A/T influences the susceptibility of Helicobacter pylori related gastric diseases in the Japanese population. Gut 2005; 54: 330-5. [ Links ]
157. Malfertheiner P, Megraud F, O´Morain C Bazzoli F, El-Omar E, Graham DY, et al. Current concepts in the management of Helicobacter pylori infection- The Maastricht III consensus report. Gut 2007; 56: 772-81. [ Links ]
158. Wong BC, Lam FK, Wong WM, Chen JS, Zheng TT, Fen RE, et al. Helicobacter pylori eradication to prevent gastric cancer in high risk region in China: a randomized controlled trial. JAMA 2004; 291: 187-94. [ Links ]
159. Lochhead P, El-Omar EM. Helicobacter pylori infection and gastric cancer. Best Pract Res Clin Gastroenterol 2007; 21: 281-97. [ Links ]
160. De Vries AC, Kuipers EJ, Raws EAJ. Helicobacter pylori eradication and gastric cancer: when is the horse out of the barn? Am J Gastroenterol 2009; 104: 1342-5. [ Links ]
161. Zullo A, Hassan C, Andriani A, Cristoffari F, De Francesco V, Ierardi E, et al. Eradication therapy for Helicobacter pylori in patients with gastric MALT lymphoma: A pooled Data Analysis. Am J Gastroenterol 2009 advance online publication 16 june 2009. [ Links ]
162. Malferthteiner P, Schulktze V, Rosenkranz B, Kauffman SHE, Ulrichz T, Novicki D, et al. Safety and inmunogenecity of an intramuscular Helicobacter pylori vaccine in noninfected volunteers: a phase I study. Gastroenterology 2008; 135: 787-95. [ Links ]

