










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.25 no.4 Bogotá Oct./Dec. 2010
Non alcoholic fatty liver disease. The new millennium pandemia
Luisa Fernanda Santos, MD (1), Geovanny Hernández, MD (2), Adriana Varón Puerta, MD (3), Óscar Beltrán, MD (4), Rafael Claudino Botero, MD (5), Gilberto Mejía, MD (6)
(1) Master of Public Health, Master of Health Administration, Hepatology Section, Department of Internal Medicine, Fundación Cardio-Infantil, Bogotá, Colombia.
(2) Second Year Resident, Department of Internal Medicine, Universidad del Rosario, Fundación Cardio-Infantil, Bogotá, Colombia.
(3) Internist, Gastro-hepatologist in the Hepatology Section of the Department of Internal Medicine, Fundación Cardio-Infantil, Bogotá, Colombia.
(4) Internist, Gastro-hepatologist in the Hepatology Section of the Department of Internal Medicine, Fundación Cardio-Infantil, Bogotá, Colombia.
(5) Internist, Chief Gastro-hepatologist in the Hepatology Section of the Department of Internal Medicine, Fundación Cardio-Infantil, Bogotá, Colombia.
(6) Hepatobiliary and Organ Transplant Surgeon, Chief of the Transplant Service, Surgery Department, Fundación Cardio Infantil, Bogotá, Colombia.
Received: 16-11-10 Accepted: 25-11-10
Abstract
Non-alcoholic liver disease (NAFLD) is the most common liver disease in affluent societies, affecting 2-8% of the general population, and it will be soon in our societies. It is generally asymptomatic or with a no specific picture of fatigue, hepatic pain or discomfort and hepatomegaly. It is suspected in cases with aminotransferase or imaging abnormalities. Liver biopsy is considered the gold standard for diagnosis. There is a close pathogenic relationship with obesity, type 2 diabetes, hyperlipidemia, metabolic syndrome and insulin resistance. Approximately 20-25% of the cases progress into cirrhosis with all its complications including hepatocellular carcinoma and the need for liver transplantation. Correction of insulin resistance with dietary measures and physical activity is generally beneficial. The efficacy of the multiple medications available remains to be demonstrated.
Keywords
Fatty liver, steatohepatitis, fibrosis, cryptogenic cirrhosis, hepatocellular carcinoma, obesity, type 2 diabetes, hyperlipidemia, insulin resistance, bariatric surgery, liver transplantation.
INTRODUCTION, DEFINITION AND CURRENT IMPORTANCE
The relationship of obesity and diabetes to liver disease (1) (obese-diabetic liver disease) was first reported over three decades ago. In 1980, Ludwig and his collaborators (2) described the condition of a series of patients, who without having a significant history of alcohol consumption, showed histopathological changes that were indistinguishable from those occurring in alcoholic liver disease. This group coined the term "non alcoholic steatohepatitis" (NASH), which has been recognized since then as one of most frequent liver diseases in the world (3). Its potential progression to cirrhosis (4), liver insufficiency (5) and hepatocellular carcinoma (6, 7) has been identified. The real importance of NASH in Colombia is unknown, but it is increasingly the reason for general consultations, and increasingly leads to cirrhosis and liver transplants.
NASHs clear association with the metabolic syndrome (X Syndrome) in nearly 80% of the patients, has helped establish its physiopathological relation to insulin resistance (8,9), obesity, hypertriglyceridemia, and life-styles in the western hemisphere (10).
The term Non-Alcoholic Fatty Liver Disease (NAFLD) was proposed by Matteoni et al. (4) with the purpose of including the whole clinical spectrum of fatty liver disease, steatohepatitis and cirrhosis. NAFLD includes four categories:
Type 1: Isolated Steatosis
Type 2: Steatosis plus lobular inflammation
Type 3: Steatosis, lobular inflammation and ballooning of hepatocytes
Type 4: Steatosis, lobular inflammation, ballooning of hepatocytes, and Mallory bodies and/or fibrosis.
These categories have prognostic implications because Types 1 and 2 remain stable while Types 3 and 4 show the potential for progression and have worse prognoses.
These entities represent a clinical and histopathological spectrum of liver damage which extends from diffuse hepatic steatosis to steatohepatitis and cirrhosis of the liver (10-30%) in the absence of alcohol, medicines, infections or toxic substances (11, 12). It is called "primary" when there is no clear cause aside from the metabolic syndrome, and "secondary" when it is clearly related to a cause (Table 1).
Table 1. Secondary causes of steatohepatitis.

Metamorphosis or non-alcoholic fatty infiltration is characterized by the infiltration of triglycerides (macro-vacuoles) and, to a lesser degree, of fatty acids (micro-vacuoles) into the liver cells without inflammation, fibrosis, liver damage, or major symptoms (13). In most cases the clinical picture remains stable, although a small percentage (<10%) may develop fibrosis and cirrhosis over a period of several decades. In its initial stage the process can be reversible when the primary cause (metabolic syndrome) is modified or interrupted. It is a matter of discussion whether a fatty liver with a minimum fatty metamorphosis (<5.0%) without inflammation or fibrosis should be considered diseased or a reversible and benign entity with no pathogenic potential. In contrast, when the initial fatty metamorphosis is associated with inflammation, cell ballooning, and Mallory bodies, the process is called non-alcoholic steatohepatitis, and there is a clear progression toward fibrosis and cirrhosis of the liver with all its complications (14).
Given that this definition rules out alcohol intake as a cause, in this review we are adopting a 140 g/week limit (ideally <20 g/day for men and <10 g/day for women).
In any case, we recommend immediate restriction of alcohol intake because of its clearly adverse effects in relation to NAFLD, and because it can facilitate interpretation of liver enzymes.
Treating metabolic syndrome and insulin resistance, modifying the diet, and increasing physical activity, all seem to be beneficial for most patients (14) including those with more advanced stages of the disease. These patients should also undergo drug therapy (15) and bariatric surgery (16) to control the process of fibrosis and progression toward chronic liver disease. However, despite innumerable clinical studies and the introduction of multiple drugs aimed at controlling liver lesions and insulin resistance, the impact of all of these therapeutic modalities has not yet been measured during the natural history of the disease (17).
The importance of this disease has come about as the result of increased longevity of the general population, improved control of viral hepatitis B and C, globalization of the economy, and changes in peoples life-styles. This disease has become one of the primary causes of chronic liver disease which generates very high costs, high morbidity rates and high mortality rates. Post-transplant recurrence (18) and a relation to cardiovascular disease have been demonstrated (13).
NAFLDs coexistence with other liver diseases, such as hepatitis B and C, modifies its natural history and makes diagnosis difficult (19, 20). Moreover, its presence among children is very worrisome (21).
EPIDEMIOLOGY
Epidemiological study of NAFLD is in the initial stages because it has only been described recently. In its beginning stages, NAFLD is asymptomatic and hard to diagnose. We do not know its real incidence, and studies of its prevalence have been based on surrogate markers for the disease such as elevation of aminotransferase or images with low sensitivity and diagnostic specificity (22). Likewise, there is no agreement about the minimum amount of alcohol or the histopathological changes required to establish a diagnosis. Considering these limitations, and accepting that it is very difficult to appropriately investigate a disease that requires hepatic biopsy as the "gold standard" for its diagnosis, NAFLD represents the most frequent liver disease in the world (23). Its prevalence is increasing due to its relation to the world wide obesity and diabetes type 2 epidemics (24). NAFLD has been reported North America (25), Australia (26), Japan (27), India (28), the Middle East (29), South America (30), North Europe (31), South Europe (32), New Zealand (32), and Southeast Asia (33). The most important studies of NAFLDs prevalence are reviewed in Table 2. They include two different general population groups. The first one encompasses very select groups of patients: patients who were referred for hepatic biopsy due to imaging changes or chronically elevated aminotransferase, patients who died in car or aviation accidents, patients evaluated for liver transplants, patients taken for autopsy after dying from different causes, and patients with morbid obesity selected for bariatric surgery. The second group includes large groups of general population of patients diagnosed with NAFLD by diagnostic imaging or by biochemical liver tests with abnormal results. This group reflects the prevalence of NAFLD in the general population more appropriately than highly selected and less representative first group. It also has the advantage of having histologically established diagnoses. This provides necessary information for understanding the stage and severity of the disease.
Table 2. Prevalence of NAFLD and NASH.
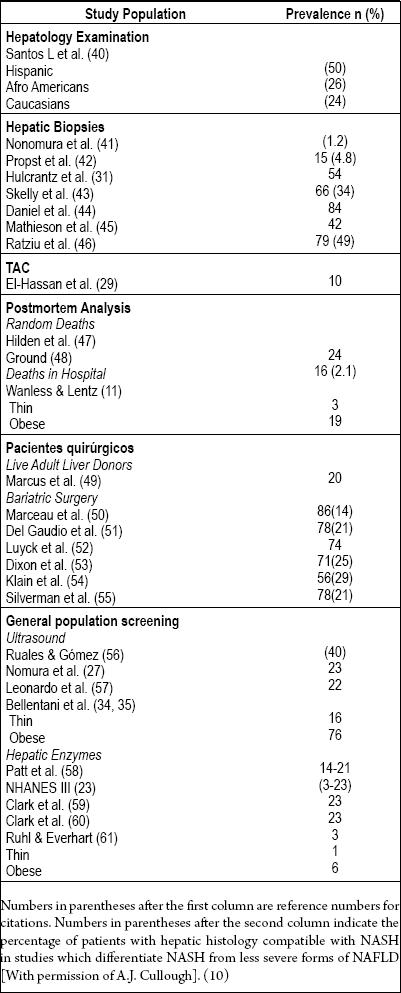
The third National Health and Nutrition Examination Survey (NHANES III) performed by the Center for Disease Control and Prevention (CDC) in the United States is noteworthy. Conducted between 1988 and 1994, it includes more than 12,000 adults drawn from the general population of the USA (23). This study diagnosed NAFLD whenever ALT, ASP or GGTP were present at elevated levels and other causes of hepatic illnesses were absent. It found prevalences ranging from 3% to 23% which were clearly related to elevated Body Mass indexes (BMIs). In addition the study showed that NAFLD is most frequent among Hispanic patients (45%) than among other patients. Prevalence among Caucasian patients was 33%, while Afro-Americans were least frequently associated with this diagnosis with a prevalence of 24%.
A second study called the Dionysos study conducted in Northern Italy from 1990 to 1992 looked at 7,000 patients who had undergone sonograms to detect fatty liver disease. 16% of the patients were thin, while 76% of these patients were obese (34, 35).
The study demonstrated that the prevalence steatohepatitis increased 46% among people of normal weight who consumed more than 30 grams of alcohol per day, and increased by 95% among obese people when they consumed this amount of alcohol.
The clear relation between insulin resistance and metabolic syndrome has generated the notion that this relation exists in all populations studied. However, recent observations among Asian (36) and south American populations clearly show that the combination of central obesity and insulin resistance seems to be more important than the combination of insulin resistance and metabolic syndrome. This suggests that anthropometric measures appropriate in the west might not be appropriate in other areas of the planet in which cases of NAFLD are found among people of normal body mass.
Numbers in parentheses after the first column are reference numbers for citations. Numbers in parentheses after the second column indicate the percentage of patients with hepatic histology compatible with NASH in studies which differentiate NASH from less severe forms of NAFLD [With permission of A.J. Cullough]. (10)
When the physician suspects a diagnosis of NAFLD, it is useful to have on hand and take into account the anthropometric data established by the World Health Organization (37) and the International Diabetes Institute (38, 39) to define overweight and obesity in the white race. Overweight is defined as BMI ≥ 25 kg/m2, and obesity is defined as BMI ≥ 30 kg/m2. Central Obesity is defined as waist circumference (WC) for men > 102 cm and waist circumference (WC) for women >88 cm or by waist to hip ratio (W:H) > 0.90 for men and >0.85 for women. For other ethnic groups the physician should consult the anthropometric definitions established for the particular group (38).
Current prevalences of NAFLD for people of normal weight vary between 1% and 24%, and between 6% and 86% for people who are obese. If we assume that 23% of the general population is obese, global prevalence of NAFLD should be between 17% and 33%. The prevalence of NASH is more difficult to estimate because studies based on liver biopsies have not been conducted in the general population. If we assume that one third to one half of the patients with NAFLD have NASH, the prevalence should be between 5.7% and 17%.
Finally, if we accept that NASH is the most frequent cause of cryptogenic cirrhosis with all of its complications including hepatic insufficiency and hepatocellular carcinoma, and that 20% of NASH cases evolve into cirrhosis within 5 to 7 years, we will understand its natural history better. As we will see in another part of this review NAFLD/NASH will become one of the primary reasons for liver transplants throughout the world by the year 2020. Moreover, the aggravation does not stop there because recurrence after transplantation is the rule. As a consequence, controlling and avoiding progressive damage to the transplanted organ requires a great deal of time and resources.
RISK FACTORS
Metabolic Syndrome
It is a good idea to define metabolic syndrome (also known as insulin resistance syndrome). This is a fundamental etiologic and pathogenic relation characterized by the following six components: arterial hypertension, central obesity, raised fasting plasma glucose, elevated triglycerides, reduced levels of high density lipoproteins (HDLs), and microalbuminuria. It is currently accepted that the presence of three of these conditions is sufficient to establish the diagnosis of metabolic syndrome, and that NAFLD is the hepatic component of this syndrome (62). A recent study of obesity showed that the risk of fatty liver disease increased exponentially with every additional component of metabolic syndrome that was found to be present. Its presence increased the possibility of NASH more than it increased that of steatosis (62).
Obesity
Although NAFLD and its most severe form NASH can occur in patients who are not obese, the majority of cases occur in people who are obese or overweight.
Some studies have shown that the average prevalence of obesity among patients with NAFLD ranges from 57% to 93% (2, 4, 32, 63). Almost all children with NAFLD are obese (64). The high prevalence of obesity in NAFLD cases could explain its association with fatty liver disease (65).
Increases in body fat content currently are given great importance now that adipose tissue is considered to have the capacity to secrete potentially toxic substances such as tumor necrosis factors (TNFs), resistin, liptin, and fatty acids which induce insulin resistance (66-69). The recent description of low levels of adiponectin and insulin resistance could be of great importance for the pathogenesis of NAFLD (70). If it is true that the body fat content is essential for the understanding of the pathogenesis of NAFLD, its distribution throughout the body seems to be even more important. In fact, it has been shown that central obesity, which favors visceral fat, is a good predictor of fatty liver disease (71), hyperinsulinism (72), and insulin resistance (72).
Diabetes Type 2
Diabetes, especially type 2, is another clearly identified risk factor for the development of NAFLD. Diabetes type 2 has been clearly related to the appearance of fibrosis (4, 11) and its progression to cirrhosis.
A recent study has established that in patients with NAFLD diabetes type 2 is a predictor independent of cirrhosis and all of its complications (73), and that it is more common in patients with NASH.
Predictors of Progression to Advanced Fibrosis
Keeping in mind that two thirds of all patients with NAFLD remain stable and never progress to development of cirrhosis, while about one third evolve toward advanced chronic hepatic disease, researchers have identified factors associated with the development of advanced fibrosis. They include histological diagnosis of NASH, diabetes (4), ages over 45 or 50 years (74), high blood pressure (53), AST:ALT greater than one (74), elevated triglycerides (46), levels of ALT (46), hepatic iron (75) and the index of insulin resistance (75).
NATURAL HISTORY
In accordance with the studies of Matteoni (4) and his colleagues, and as mentioned earlier, NAFLD is a spectrum of entities. Steatosis alone is Type 1. Steatosis plus inflammation is Type 2. Steatosis, lobular inflammation, and hepatocyte injuries (ballooning degeneration) together define Type 3, while steatosis and ballooning degeneration with or without Mallory bodies and/or fibrosis define Type 4. NAFLD does not necessarily evolve in ascending order of types. There exist no age differences between Types 1 and 2 which are stable, and Types 3 and 4 which are progressive and grouped together as NASH. This suggests that rather than representing different stages of evolution of one process, these Types represent different entities. This indicates that Type 1 and Type 2 patients are capable of controlling the process and remaining stable, while Type 3 and Type 4 patients develop in the direction of hepatocyte injuries and progression to cirrhosis. 5 to 7 year clinical and histopathology follow-up studies (76) have shown that the Types 1 and 2 remain stable without great changes or complications. Less than 3% of these cases progress to cirrhosis. In contrast, 60% of patients with the progressive types (Types 3 and Type) remain stable, but 20% develop fibrosis and 20% develop cirrhosis with all of its complications including hepatocellular carcinoma. Also, histological follow-up studies have indicated that there is a substantial risk of progression when the initial biopsy sample shows changes in Types 3 and 4 (NASH). One study of great importance is that done by Powell et al. (26) which followed 13 patients for an average of 4.5 years. It showed progression to cryptogenic cirrhosis with disappearance of the histological changes typical of NASH. In this way they were able to identify several risk factors which predict progression (see before). It is necessary to indentify risk factors which will allow us to predict the natural history of the process from its initial stages.
One frequent fact in clinical practice is that the absence of hepatic complications related to metabolic syndrome causes confusion in which hepatic encephalopathy is mistaken for depression, hepatopulmonary syndrome for other pulmonary problems related to obesity, edema caused by cirrhosis for cardiac failure, hypersplenism for immune thrombocytopenic purpura (ITP), varicose bleeding for peptic ulcer disease, and cirrhotic ascites for malignancy. By the same token, treatment of metabolic syndrome with angiotensin-converting enzyme (ACE) inhibitors, aspirin and other antithrombotics worsens retention of liquids and adversely affects the already compromised coagulation.
Despite the fact that cirrhosis is a frequent cause of death among patients with NAFLD and diabetes type 2, it has been shown that mortality due to cardiovascular events actually masks the real mortality rate due to hepatic causes (77, 78). 19% of deaths among these patients are due to cardiac events, 16% to cerebral vascular events, 13% to renal failure, and 6% are hepatic. Finally, Ayata and his colleagues (79) demonstrated through a study of hepatic explants that cryptogenic cirrhosis is related to NASH in 30% to 70% of all cases. Furthermore this has also been shown through a careful retrospective follow-up of patients with NAFLD in which obesity and metabolic syndrome were much more frequent than in other types of cirrhosis. They were present for long periods of time but then disappeared or were minimized under the influence of loss of body fat related to treatment or the development of cirrhosis. Similarly family backgrounds of cirrhosis due to unknown causes, obesity, and metabolic syndrome are also frequently found.
It is speculated that diminished blood flow in the cirrhotic nodules consequent to transjugular intrahepatic portosystemic shunts (TIPS) (80) and the capillarization of the sinusoid which significantly affects the flow of lipoproteins to the liver are the reasons that the histopathological changes typical of progression toward cirrhosis disappear. The prognosis for cirrhosis among obese patients is much worse than the prognosis for NASH and is similar to that for hepatitis C. This is in part because of the greater age of patients and the extinction of the parenchyma which is observed in cirrhosis (81). The majority of studies demonstrate that women are predominantly affected by cases of cryptogenic cirrhosis. Finally a relation between NAFLD and hepatocellular carcinoma has been demonstrated to occur primarily among men (82). Moreover, this relation had been demonstrated to augment the occurrence of malignancies among obese patients with alcoholic hepatitis and NASH (83).
PATHOGENESIS
As mentioned earlier, NAFLD is considered to be a hepatic manifestation of metabolic syndrome or insulin resistance which is associated with other clinical expressions of the syndrome such as obesity, diabetes type 2, dyslipidemia and arterial hypertension (84).
Steatosis development mechanism underlying progression to steatohepatitis is still not completely understood. Probably it depends on the presence of many factors within the context of genetic predisposition to the condition. In this complex scenario the "two hits hypothesis" was posited in 1998. The first hit induces an accumulation of fat in the liver, while the second hit of oxidative damage causes progression to non-alcoholic hepatitis as observed in Figure 1 (85). Nevertheless, recently this theory has been modified to suggest that the mechanisms that cause progression also induce steatosis. Oxidative stress, cytokines and intestinal flora produce steatosis, necroinflammation and fibrosis occur jointly rather than sequentially. Free fatty acids initiate apoptosis in hepatocytes and then endoplasmic stress results in steatosis, oxidative stress and apoptosis. In other words there are several "first hits" that result in steatosis and hepatocellular damage in susceptible individuals. Steatosis should be considered to be a temporary response adapted to stress and not as the first hit in the progression of the disease (85, 86).
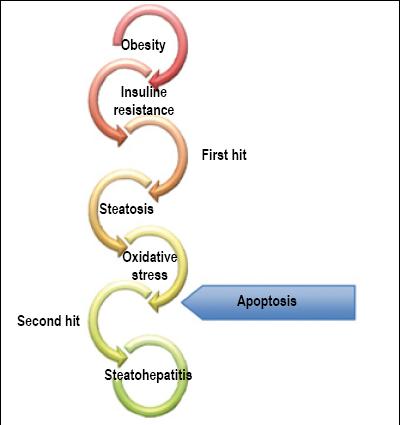
Figure 1 Pathogenesis: Two-hit hypothesis (Personal Archive)
FACTORS RELATED TO PATHOGENESIS
Genetic Predisposition
Despite the high prevalence of risk factors for insulin resistance, only a fraction of those individuals develop NAFLD and a minority of them progress to steatohepatitis and its complications. This suggests that genetic predisposition and environmental factors play very important roles. There are a number of genes which are not only related to accumulation of fats but are also related to other mechanisms including oxidation, oxidative stress, adipokines and their receptors, and cytokines and their receptors (87).
Increased production and/or accumulation of lipids at the hepatocellular level
Pathogenic mechanisms which produce an accumulation of triglycerides and free fatty acids in the hepatocytes can easily be explained by taking into account the normal cycle of free fatty acids between adipose tissue and the liver. Steatosis is the result of a defect in the metabolic process of fats (Figure 2) (88).
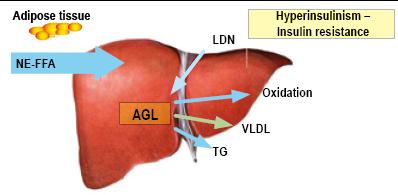
Figure 2. Role of free fatty acids in NAFLD pathogenesis. (AGL) FFA: Free Fatty Acids. NE-FFA (AGL-NoE) Non-esterified free fatty acids. DNL (LDN): De novo lipogenesis. TG: Triglycerides. LDLP (VLDL): Low density lipoproteins (Personal Archive)
- Hepatic capture of free fatty acids from adipose tissues increases (60% to 80%).
- Lipogenesis increases (5% in healthy patients increasing to 26% in patients with NAFLD).
- Free fatty acids increase by 15% due to diet.
- Beta oxidation of free fatty acids is insufficient to generate ATP. This is associated with a deficiency of dehydrogenated acyl-CoA (valproic acid (VPA), chronic use of aspirin, vitamin B5 deficiency or excessive alcohol consumption.)
- Diminished synthesis and secretion of very-low-density lipoproteins (VLDL) which is medically associated with tetracyclines and amiodarone.
Insulin resistance
Insulin resistance plays a key role in the development of hepatic steatosis and steatohepatitis. Insulin resistance is defined as the condition in which larger concentrations of insulin are needed to achieve a normal metabolic response, or the condition in which normal concentrations of insulin fail to achieve a normal metabolic response. Insulin stimulates the capture of glucose by the peripheral tissues (especially the muscles) and suppresses its production in the liver (89).
Insulin resistance first develops peripherally in the adipose tissue. In adipocytes insulin inhibits lipase which is sensitive to the hormone, preventing lipolysis of triglycerides and liberation of free fatty acids. Insulin resistance is generated by a change in the insulin receptor which diminishes glucose capture by muscles and by adipocytes, and which suppresses lipolysis resulting in increased synthesis of triglycerides. Insulin resistance in the liver alters glycogenesis by increasing gluconeogenesis and glycogenolysis resulting in increased capture of free fatty acids (90).
Insulin resistance promotes accumulation of fat not only through increasing free fatty acids in the liver, but also through hyperinsulinism by stimulating hepatic lipogenesis. In addition it promotes the progression of steatohepatitis and fibrosis by inducing oxidative stress, stimulating proliferation of hepatic stellate cells (HSCs) and stimulating secretion of extracellular matrix (ECM) (91).
Oxidative Stress and Mitochondrial Dysfunction
Free fatty acids are substrates and induce microsomal lipoxygenase of cytochrome p-450 producing hepatotoxic free radicals. Mitochondrial Beta oxidation of free fatty acids, as the most important pathway for oxidation, results in increased formation of free radicals, and increased hepatocellular damage and fibrosis. Electronic microscopes reveal mitochondrial structural abnormalities among patients with steatohepatitis which do not appear among patients with fatty liver disease secondary to increased lipid peroxidation. Some researchers have postulated that, in the absence of mitochondrial defects, insulin resistance will only generate non-alcoholic fatty liver disease (92).
Apoptosis
Recognition of hepatic apoptosis as a key pathway for cellular death and damage among patients with steatohepatitis has surged. Levels of cytokeratin 18 (CK-18) fragments are used to measure it. These levels are normal among patients with NAFLD, are high among patients with steatohepatitis, but diminish after bariatric surgery. CK-18 is an intermediate intrahepatic filamentous protein which is cleaved by caspase enzymes during apoptosis. It correlates well with inflammation and fibrosis in other pathologies such as hepatitis B and hepatitis C (93, 94).
Cytokine and adipokine interrelationship
Cytokines and adipokines are mediating molecules involved in the physiopathology of many diseases. Adiponectin, leptin, tumor necrosis factor-alpha (TNF-A) and interleukin-6 (IL-6) are associated with visceral obesity and play an important role in modulating the action of insulin in the inflammatory cascade in patients with NAFLD (84, 95).
Adiponectin is a hormone which is exclusively excreted by adipose tissue. It has beneficial effects on the metabolism of lipids including increasing plasma lipid clearance and increasing beta oxidation of fatty acids in the muscles. Its levels are inversely associated with severity of steatosis and play a protective role in relation to insulin resistance.
Leptin induces dephosphorylation of the insulin receptors which makes hepatocytes more resistant to insulin. In addition, it promotes inflammation and fibrogenesis. Its levels are high among obese patients and among those with NAFLD.
Resistin is a protein derived from adipocytes which can play an important role in insulin resistance. Its over-expression in animals increases glucose intolerance and hyperinsulinism and alters suppression of free fatty acids.
TNF-A and IL-6 have elevated levels in patients with insulin resistance, steatohepatitis and very severe fibrosis (Figure 3).
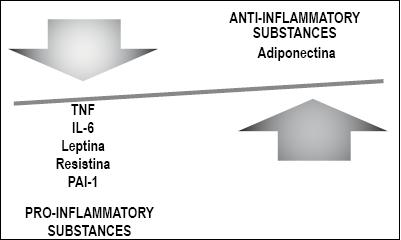
Figure 3. Pathogenesis: Adipokines and Cytokines. (Personal Archive)
Other adipocytokines whose roles in NAFLD have been investigated are adipocytes protein united with fatty acids, apelin (ligandin for the angiotensin receptor) and vaspin.
Bacterial Overgrowth
Intestinal bacteria are associated with the second strike or hit because they are a potential cause of oxidative damage. One study observed that bacterial overgrowth was greater among patients with steatohepatitis than among control patients (96). Increased production of respiratory ethanol has been documented among patients with steatohepatitis. This increased the risk of hepatic damage among obese patients after bariatric surgery, but this improves with the use of metronidazole after surgery. This suggests that the bacterial overgrowth is a risk factor for severity of hepatic illnesses among obese patients. One theory posits that bacterial overgrowth contributes to the pathogenesis of NAFLD by increasing endogenous production of alcohol, acetaldehyde and cytokines by way of the exposition of lipopolysaccharides (LPS). Nevertheless, the evidence for this theory is still weak (97).
Mechanisms under investigation (84)
- Endogenous endocannabinoids and their CB1 and CB2 receptors
- Toll-like receptor TLR-4
- Serotonin
- Retinol-binding protein 4 (RBP4) and its relation to insulin resistance
- Vitamin D3
- Dehydroepiandrosterone (DHEA)
CLINICAL MANIFESTATIONS AND DIAGNOSIS
Although NAFLD patients can be of any race or ethnic background, there is a clear predilection for NAFLD in the white race and among Hispanics during the 4th and 5th decades of life (40, 98). Some early studies have suggested a predominance of women among NAFLD patients, but more recent studies have shown a predominance of male patients (99). The majority of patients present clear associations with one of the following factors: obesity (10% to 40%) (100), diabetes mellitus (21% to 75%) (99), hyperlipidemia (21% to 83%) (99), and arterial hypertension (15% to 68%)(101). Combination is associated with greater severity of NAFLD (53). Nevertheless, NAFLD can occur in thin individuals who have none of these other disorders. The majority of these cases are insulin resistant or have clear family histories of insulin resistance (25). This entity is generally asymptomatic, so the majority of cases of discovered incidentally to other causes because of high levels of aminotransferase. Some patients consult a physician because they do not feel well or have a sensation of heaviness in the right upper quadrant induced by distension of Glissons capsule caused by excessive hepatic fat (25). The patients physical examination is generally normal, and classical stigmas of cirrhosis are rarely encountered. Some female patients present hirsutism and acne as a result of endocrinal disorders of ovarian polycystic syndrome or lipodystrophy. One common finding is acanthosis nigricans clearly related to insulin resistance. About 30% of NAFLD cases experience liver pain upon deep palpitation (25).
Laboratory Tests
Hepatic tests generally show elevated levels of aminotransferase (less than 4 times the normal limit), alkaline phosphatase and gamma-glutamyltransferase (GGTP). In contrast to alcoholic hepatitis, ALT usually predominates in the initial phases of NAFLD with AST/ALT <1. Aminotransferase activity is not correlated with the severity of the illness, and it is not rare to encounter cirrhotic patients with normal aminotransferase. Bilirubin and prothrombin time only affect very advanced cases of cirrhosis. Hyperlipidemia can be observed in from 21% to 83% of cases, the majority of which also have hypertriglyceridemia. Since the majority of these patients also have diabetes type 2, glycemia and elevated levels of hemoglobin A1c are found. Ferritin levels are elevated in from 40% to 62% of cases, but the level of hepatic iron is normal. It is common to find positivity to low quantities of antinuclear antibodies (ANAs) without necessarily encountering autoimmunity. Although some earlier studies have suggested high frequencies of human hemochromatosis protein (also known as HFE protein) in these patients, more recent studies have not supported this finding. Finally, coexistence of NAFLD with other hepatic diseases such as viral hepatitis, primary biliary cirrhosis, autoimmune hepatitis, hemochromatosis and alpha 1-antitrypsin (A1AT) deficiency is not rare. For this reason it is necessary to completely rule out these other entities when studying patients with NAFLD.
Now non-invasive elastography tests are available for detecting fibrosis. They include FibroTest® from BioPredictive in Paris, France; FibroSURE® from LabCorp in Burlington, VT (USA) and FibroScan® from Echosens in Paris, France. They measure hepatic rigidity using sound probes which use a transducer to capture the echoes rebounding from the tissue. The signal is then sent to a computer which produces a number which correlates very well with the degree of fibrosis. These tests are promising, and may replace biopsies in the future. Nevertheless, their limitations lie in their ability to detect the extremes of the spectrum of minimal fibrosis and advanced fibrosis rather than in detecting intermediate grades. Elastography also present many falsely positive results in obese patients. Some centers use them simultaneously with initial hepatic biopsies, after which follow ups are done solely with elastography which is very convenient for patients. Nevertheless, until research can validate these tests they should be considered experiment and thus are not currently recommended.
Diagnostic Imaging
Methods based on imaging offer various advantages over biopsies and histopathological analysis for diagnosing NAFLD. Imaging is not invasive, plus it allows study of other hepatic abnormalities including hepatocellular carcinoma in high risk patients. The simplest method for detecting and characterizing hepatic steatosis is ultrasound (echography).
The parenchyma of the normal liver has a homogenous echotexture with echogenicity equal to or slightly higher than the renal cortex and spleen. The intrahepatic blood vessels, including the portal vein and the hepatic veins, are easily identifiable. Hepatic steatosis produces "brilliants" in the hepatic parenchyma where the echogenicity becomes greater than that seen in either the renal cortex or the spleen (Figure 4). Degrees of hepatic steatosis are light, moderate and severe. Light hepatic steatosis is characterized by increased hepatic echogenicity with clear definition of the walls of the portal vein and the hepatic veins. Moderate hepatic steatosis is characterized by increased echogenicity with partial blurring of the walls of the portal vena and the hepatic veins. Severe hepatic steatosis has increased hepatic echogenicity combined with a loss of the posterior hepatic contours (102).
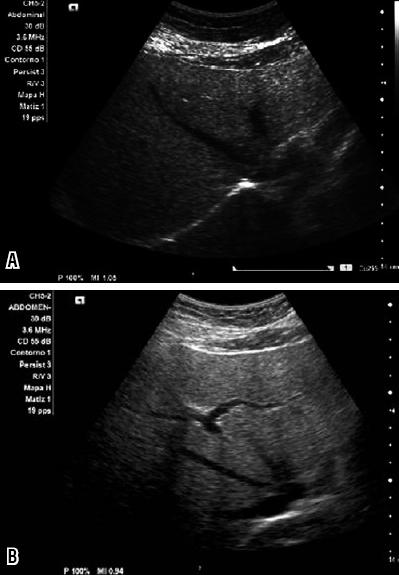
Figure 4. Sonogram images of various patients. Observe the increased echogenicity of the hepatic parenchyma resulting from fat infiltration (Personal Archive)
Despite the benefits of ultrasound, including easy access, absence of ionizing radiation and low cost, it has important limitations. These include a limited field of study, no quantitative measurements, poor sensitivity for differentiating between hepatic steatosis caused by fibrosis and cirrhosis and the fact that this method depends on the quality of the operator and the equipment. Together these limitations constitute an impediment for its generalized use for evaluation of fatty liver disease.
High resolution computed tomography (HRCT) is a technological advance which has substantially increased the use of CAT scans for studying routine hepatic illnesses. High resolution CT scans without the use of a contrast agent are considered to be the best method for evaluating hepatic fat since it permits measurement of the attenuation of the liver in Hounsfield units (HUs). The Hounsfield scale measures attenuation based on physical characteristics of x-ray penetration of tissues. With HRCT the hepatic fat content can be evaluated subjectively by visual inspection, and objectively by selecting a region of interest (ROI) and measuring values of attenuation within that range. Hounsfield units within a hepatic ROI compare to those of the ROI of the spleen. The normal liver is more highly attenuated than is the spleen (Figures 5a and 5b). When the livers attenuation is less than that of the spleen, a diagnosis of hepatic steatosis can be considered. Various studies have confirmed that this method has high sensitivity (88% to 95%) and high specificity (90% to 99%).
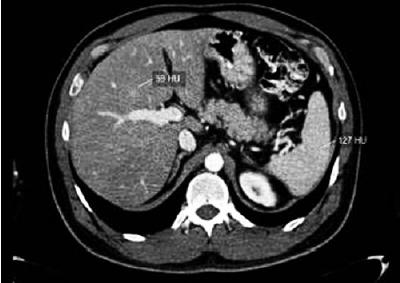
Figure 5a. This CT scan shows the decrease in the coefficient of attentuaiton of the hepatic parenchyma as measured by Hounsield Units (HU). To diagnose fatty liver disease the difference between the HU measure of the spleen and that of the liver should be greater than 20 HU. (Personal Archive)
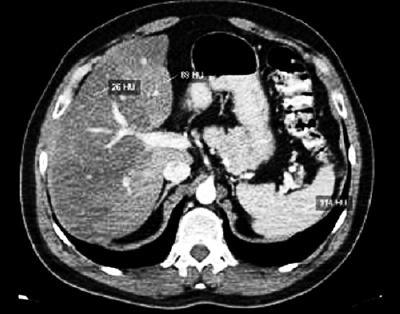
Figure 5b. This CT scan shows the geographic distribution of hepatic fat infiltration in which part of the hepatic parenchyma is compromised while other parts are not (Personal Archive).
Another advance in CT scanning technology is the use of two x-ray tubes or two rows of detectors. With this technology, values for liver attenuation range between 60 and 65 HUs. Infiltration of fat is diagnosed when attenuation of the liver is less than 48 HUs. It has been found that attenuation of 40 HUs represents changes in fat content of approximately 30% (103).
Magnetic resonance imaging (MRI) is one of the most sensitive methods for studying the liver. Two techniques are used: chemical displacement and spectroscopy.
Chemical displacement: The protons of hydrogen atoms, which are used in MRIs, normally have spinning motions, however the protons of hydrogen atoms in water molecules and fat molecules spin at different velocities. If the protons of water molecules and fat molecules are looked at from the same side (phase), their signals add together (Figure 6a), but if they are looked at from opposite sides (out of phase), their signals cancel each other out and the image appears to be black (Figure 6b). In a normal liver there are insufficient fat molecules to produce this effect. Consequently, out of phase images of the liver look the same as in phase images. However, if the liver is fatty, the effect will be produced and the out of phase image will appear black. The technique of chemical displacement has the advantages of being technically simple, of covering the entire liver, of being more specific, and of being radiation free. It has the disadvantage of being unable to quantify the amount of fat (104).
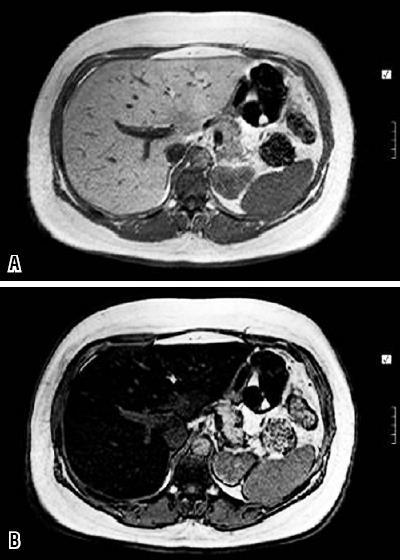
Figure 6. MRI of fat infiltration. These images were obtained from a chemical displacement sequence. Figure 6a. Normally the liver and the spleen do not change in signal intesnity. Figure 6b. When there is fat infiltration in the liver it looks black when images are out of phase. The spleen can be used for comparison. (Personal Archive)
Spectroscopy: Spectroscopy makes it possible to see the composition of organs and to be able to quantify the amount of fat present, including very small quantities in the hepatic parenchyma. When this method is used a spectrum graph is generated with various peaks indicating the composition of the tissue examined. It has the advantages of being able to determine the absolute concentration of hepatic lipids and of being able to detect small quantities.
Hepatic biopsies
Histopathological analysis continues to be the gold standard for diagnosis since none of the other diagnostic methods (including imaging techniques, non-invasive methods such as elastography, and blood tests) have the ability to detect stages of the disease or the presence of other concomitant entities such as hepatitis C, Wilsons disease, or µ1 antitrypsin deficiency. Despite its benefits, hepatic biopsy is an invasive procedure which is uncomfortable for patients and which can cause problems such as post-biopsy pain and even serious complications such as bleeding, perforation of an organ, and death. Studies have reported that 0.01% of hepatic biopsies result in fatalities (105).
Also, the possibility of a sample error could generate confusion in the diagnosis or in measuring the severity of the condition. Another reason to question the usefulness of biopsies is the lack of truly effective treatments which can alter the natural history of the disease although some patients change their habits after the experience of the biopsy and after learning its results.
HISTOPATHOLOGICAL CHANGES
As we stated initially, NAFLD includes a spectrum of severity extending from the metamorphosis of liver fat with minimum inflammation to steatohepatitis and cirrhosis. Although histopathological changes in alcoholic steatohepatitis and NAFLD are indistinguishable, in general the severity of these changes is much greater in alcoholic steatosis than in NAFLD, and some changes such as alcoholic hyaline necrosis, cholestasis and ductal proliferation are specific to alcoholic steatosis. For this reason the clinical physician and not the pathologist should establish with certainty, based on all of the available evidence, the real etiology of the problem. In the following section we will discuss which histopathological changes are typical in NAFLD. The clinical physician should be able to recognize these typical changes and discuss them with a pathologist without fear (106).
Hepatic Steatosis
Hepatic steatosis is characterized by an accumulation of triglycerides and fatty acids in the hepatocytes. The principal observations seen in a sample stained with hematoxylin and eosin (H&E staining) under a light microscope are macrovesicular inclusions which displace the cellular nucleus to the periphery of the cell (Figure 7). To a lesser degree microvesicular fat inclusions can be observed surrounding the nucleus. In the initial stages (Types 1 and 2) the steatosis is located principally in zone 3 of the hepatic lobule. It is associated with minimal inflammation and is completely reversible within a few weeks. The extension of steatosis can be estimated with the scoring system recommended by Brunt et al. (12). It has three grades. Grade 1 is characterized by fat globules in less than 33% of the hepatocytes. Grade 2 is characterized by fat globules in 33% to 66% of the hepatocytes, and Grade 3 is characterized by fat globules in more than 66% of the hepatocytes.
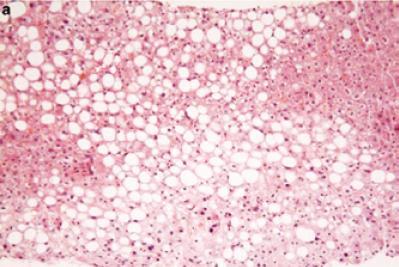
Figure 7. NAFLD: Fatty Liver (Macrovesicular and microvesicular globules of fat). Note the absence of inflammation and fibrosis. (Hematoxilin & Eosin stain, 100 x magnification). [With the permission of E. Brunt] (109)
When most of the steatosis is microvesicular, the physician should think about other possible causes including fat metamorphosis due to pregnancy, Reyes syndrome, and toxicity due to valproic acid or antiretroviral drugs. Hepatic biopsies processed by freezing frequently show an excess of microvesicular fat which, in the case of a liver donor, is not a contraindication for the procedure.
Steatohepatitis
As the name implies, steatohepatitis signifies the presence of fat and injury to the hepatocytes. It is associated with various degrees of inflammation of the hepatic parenchyma and by ballooning hepatocyte lesions. These hepatocytes have increased size due to an excess of water and are paler than the background. They are considered to be reversible, but necrosis, hepatocyte apoptosis (Mallory bodies) and megamitochondria are irreversible (11, 12) (Figure 8). The majority of authors on this topic consider the presence of fat, ballooning and hepatocyte injury to be the minimum histopathological changes required for establishing a diagnosis of NASH which evolves into advanced fibrosis (Figure 9) and cirrhosis in between 25% and 30% of these cases.
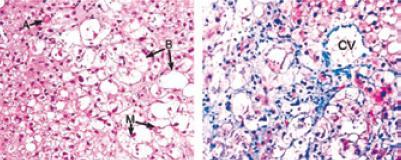
Figure 8. NASH: Arrow A shows a hyaline body. Arrow B shows ballooning hepatocytes. Arrow M shows Mallory bodies. CV shows the chicken wire pattern of fibrosis around the central vein. (Hematoxilin & Eosin, 100 x magnification y Tricromo de Masson, magnification X 100). [With permission of B.A. Neuschwander-Tetri] (110)
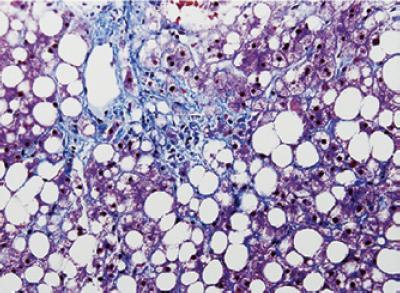
Figure 9. Non-alcoholic steatohepatitis (NASH): Note the combination of infiltration of fat, inflammation and peri-central and pericellular fibrosis. (Masson trichrome, 100 xMagnification). [With the permission of K. Law] (111).
Fibrosis and Cirrhosis
NASH fibrosis emulates alcoholic hepatitis in beginning in zone 3 (centrilobular). It is characteristically pericellular and perivenular in distribution, simulating a netting of chicken wire.
In the pediatric population fibrosis is characteristically periportal for reasons unknown. Although H&E staining of a sample may show fibrosis, either Masson trichrome stain (van Glisson) or Sirius red stain is essential for estimating the severity of fibrosis (107). With the passage of time and the progression of the illness fibrosis extends to other regions of the hepatic parenchyma causing gradual loss of normal architecture as it is replaced by regenerative nodules surrounded by bands of fibrosis (Figure 10).
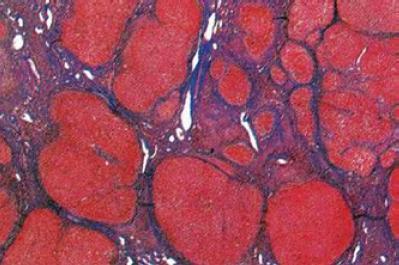
Figure 10. NAFLD: Cirrhosis. Note the absence of histopathological changes typical to NASH and the presence of regenerative nodules surrounded by fibrosis partitions. (Masson trichrome, 100 x magnification) (Personal Archive)
For reasons which are still not known, once cirrhosis has been established, the characteristic histopathological features of NAFLD disappear making it impossible to make a specific diagnosis. 30 years ago, before its relation with NAFLD was understood, the term cryptogenic cirrhosis was proposed. Now it has been replaced by the more appropriate expression: NASH related cirrhosis. In order to evaluate the severity of inflammation and fibrosis, and to be able to conduct multicentric therapeutic studies, Brunt et al. and more recently another group of expert pathologists (12, 108), have proposed classifications which all up-to-date pathologists should use to study and report cases of NAFLD (Tables 3 and 4).
Table 3. Necroinflammatory activity grades.
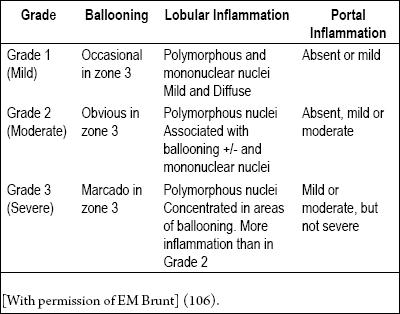
Table 4. Fibrosis stages.

Current management and therapeutic perspectives
Treatment objectives (112-114)
1. Correct insulin resistance and hyperinsulinism.
2. Reduce fatty mass, in particular visceral obesity.
3. Prevent or reverse hepatocellular damage induced by lipotoxicity.
Non-pharmocological measures
Weight loss
- No minimum required weight loss has been established to date for improving steatohepatitis
- A 5% to 10% weight loss can normalize aminotransferases.
- A 9% weight loss can improve steatosis, but not fibrosis
- The consensus weight loss recommendation for overweight and obese patients is 7% (115)
Dietary Measures (112, 116)
- No optimum diet has yet been established for weight loss. One recent study compared four types of diets. Independent of the macronutrients of each diet, all four were beneficial when adhered to. Only 15% of patients loss more than 10 kg. Adherence to diets decreases after the first months and the majority of patients regain weight lost (117).
- Nutrients which worsen NAFLD include corn syrup (fructose and glucose), non-dietetic or carbonated beverages, Trans fats (hydrogenated vegetable oils such as contained in the majority of canned foods) (118).
- Recommended nutrients include carbohydrates with low glycemic indices which are high in fiber such as fruit, vegetables, legumes and grains. Monounsaturated fatty acids such as olive oil are preferable to saturated fatty acids such as those found in meats, fats and dairy products. Polyunsaturated fats such as found in fish and nuts should be increased.
- There is no evidence that abstinence from alcohol benefits NAFLD patients. Some studies suggest that one glass of wine helps protect against insulin resistance (119).
Exercise (120)
- Only 20% to 33% of NAFLD patients are physically active.
- The consensus recommendation of various societies derived from studies regarding prevention of diabetes is for 150 minutes of moderate exercise (brisk walking) weekly, adding 75 minutes weekly of running and muscular strengthening (anaerobics) twice a week (121).
- Although these recommendations should be individualized, the patient should be stimulated to conduct any physical activity, even if it is limited, because it will always be beneficial to the patient compared to a completely sedentary life.
- Four weeks of aerobic exercise decreases hepatic fat and visceral obesity even when there is no change in weight or modification of diet. Exercise for more than 3 months, independent of weight loss, increases cardio-respiratory functioning, improves insulin resistance and increases hepatic enzymes.
Bariatric surgery
This surgery can be beneficial for those patients whose BMIs are over 40, or whose BMIs are over 35 when they also have other associated illnesses. Loss of weight (34kg +/- 17 kg) improves metabolic syndrome and 82% of steatosis cases. Other benefits include improvements in sleep apnea, depression, infertility and quality of life. A recent metaanalysis showed improvement of hepatic histology after bariatric surgery (Gastrojejunostomy, Roux-en-Y anastomosis, and gastric banding). Nevertheless, a systematic review of the Cochrane collaboration concluded that bariatric surgery is not recommendable for patients with NAFLD given the lack of randomized controlled studies of the risks and benefits of this type of surgery (122-124).
A recent publication of the European Association for the Study of the Live (EASL) systematically recommends liver biopsies for all these patients given that hepatic damage is asymptomatic even when there is advanced fibrosis or cirrhosis (112).
Pharmacological therapy for associated illnesses (112, 125)
- Steatosis. Pharmacological therapy is not recommended for NAFLD. Efforts should be directed towards preventing extrahepatic complications of steatosis and metabolic syndrome, especially cardiovascular risks.
Annual clinical follow-up appointments are recommended to monitor aminotransferases and to monitor markers of insulin resistance using homeostatic model assessment (HOMA). If aminotransferase levels are inexplicably high and/or metabolic factors have worsened, a hepatic biopsy is justified within 5 years of the initial biopsy.
- Steatohepatitis. The objective of therapy is to prevent progression from fibrosis to cirrhosis. Fibrosis stages 0 and 1 do not require pharmacological therapy. Therapy specifically centered upon the liver is recommended for patients with steatohepatitis and intermediate fibrosis (Stage 2), for patients with increased histological activity and risk factors for fibrosis (patients over 50 years old, diabetes, arterial hypertension and insulin resistance), and for patients with Stage 3 fibrosis (who probably have cirrhosis).
In Stage 2 and 3 patients hepatic biopsies may be repeated to evaluate progression towards cirrhosis after non-invasive methods have been completely validated. In addition these patients should be monitored for the appearance of esophageal varices and hepatocellular carcinoma.
Pharmacological therapy centered on the liver (112, 113)
Currently there are no approved therapies for steatohepatitis. Those which are available are considered to be only for experimental use for this pathology.
Insulin sensitizers. Metformin and Thiazolidinediones (TZDs) are insulin sensitizers. Since 80% of patients with steatohepatitis have characteristics of insulin resistance, it is biologically plausible to treat them for insulin resistance.
Metformin and TZD are associated with normalization of aminotransferases in 50% of cases. This decreases the level of steatosis (evaluated by ultrasound and spectroscopic MRI) and partially improves necroinflammation but not fibrosis after one year of treatment. Continued treatment does not produce continuing improvements, but when it is suspended the histology and the level of aminotransferases return to their initial state. Because of its cardiovascular effects, rosiglitazone was recently taken off of the market.
Lipid-lowering drugs (LLDs)(Hypolipidemic agents, or antihyperlipidemic agents). Fibrates, statins and omega 3 improve aminotransferase levels but without having any advantages over changes in lifestyle. Fenofibrates PPAR-Y activity (receptors which activate proliferation of peroxisomes) is promising for NAFLD patients, although it has not yet been tested in randomized studies.
Antihypertensives. Antihypertensives effects on hepatic illnesses are uncertain. One pilot study of losartan showed moderate effects on hepatic histology. These drugs are recommended only for patients with NAFLD and arterial hypertension.
Antioxidants. Oxidative stress plays an important role in the theory of the second strike or hit. For this reason use of antioxidants to treat NAFLD is rational. One recent randomized controlled study by the NASH Clinical Research Network which compared pioglitazone, placebos, and Vitamin E showed improvement of steatohepatitis without any increase of fibrosis among patients who received 800 IUs of Vitamin E daily for two years.
Other antioxidants such as betaine and acetylcysteine have not shown convincing results.
Cytoprotective agents. Ursodesoxicolic acids hepatoprotective effects have led to several studies of its use in high doses and combined with Vitamin E, but so far the results have been contradictory.
Anorectic medications. Orlistat and sibutramine have no shown any beneficial effects for hepatic histology, although they may increase the effects of behavioral therapy.
Recently sibutramine was taken off the market because of its cardiovascular effects, while Orlistat has been associated with isolated cases of fulminant hepatic failure.
New Agents. Studies are currently being done on pentoxilin (an inhibitor of alpha TNF), caspase inhibitors, incretin analogs (exenatide and sitagliptin), and second generation sulfonylureas (repaglinide and nateglinide). In the future these might become options for treating NAFLD.
In conclusion, although there has been a decade of research and clinical studies, no pharmacological intervention has yet shown itself to be unquestionably effective for management of NAFLD and steatohepatitis. Lifestyle changes continue to be the fundamental treatment approach, with bariatric surgery as an option for patients who are extremely obese. The available information suggests that the use of Vitamin E for one or 2 years is recommendable for patients with steatohepatitis. Until new information becomes available, it might be suggested that patients whose cases are developing should be treated with pioglitazone or high doses of ursodesoxicolic acid.
NAFLD, NASH, AND HEPATIC TRANSPLANTS
NAFLD and NASH have given rise to an ironic combination of adverse effects for liver transplants. Although they increase the necessity for more donors, they simultaneously have decreased the number of donors available. By the same token it is clear that hepatic steatosis and the comorbidities associated with it have a direct impact on post-surgical results, patient survival, survival of the allograph and the specific management of these patients in the short, medium and long term (126).
Donors with fatty livers
In 1991 Adam et al. (127) identified primary dysfunctions of inserts in 13% of patients when biopsies had confirmed steatosis of greater than 30% compared to only 3% among those patients with no steatosis. As a result the majority of medical centers performing transplants avoid the use of allographs with histological evidence of macrosteatosis of more than 30%. Nevertheless, when the increased demand for organs and the scarcity of donors are taken into account, each case must be evaluated independently, considering the characteristics of the recipient, the Model for End-Stage Liver Disease (MELD) score, and prior results from the use of donors with fatty liver disease. On the other hand when a hepatic biopsy confirms a diagnosis of NASH, it is clear that such an allograph cannot be transplanted (128).
On additional variable which should be controlled is cold ischemia time. This is the period of time during which the allograph is without perfusion. Many studies have demonstrated that prolonged cold ischemia times lead to numerous complications with allographs in general, but, especially with fatty liver allographs.
NAFLD and NASH as Reasons for Hepatic Transplants
As noted above, the diagnostic problem is that steatosis typically disappears with the onset of cirrhosis suggesting that many of the cases classified as cryptogenic might in fact be late detected NAFLD/NASH cases (129). Various studies have demonstrated that when diagnostic criteria are used appropriately 2/3 of all cases characterized as cryptogenic cirrhosis turn out to really be cases of NAFLD/NASH (130).
A recent study has shown evidence that the risk of hepatocellular carcinoma (HCC) in NASH patients increases among patients over 50 years of age, and that male patients are predominant among NAFLD/NASH patients (131). For this reason follow-up examinations using imaging and alpha-fetoprotein tests are recommended every six months for these patients, as well as for patients with cirrhosis from other causes. HCC currently is considered to be an indication for hepatic transplant, as are the classical complications of cirrhosis.
NASH will be used substantially more frequently as an indicator for hepatic transplants in the future to the extent that diagnostic criteria become more specific, the obesity epidemic worsens, the number of cases of cirrhosis due to hepatitis C decrease and the complications related to pre-transplant metabolic syndrome become well controlled.
Data from the United Network for Organ Sharing (UNOS) confirms the rapid increase in the number of cases of hepatic transplants as a result of NASH. In 1996 0.1% of transplants were performed due to NASH, but in 2005 the proportion had reached 3.5% (132). NASH is projected to become the most common reason for hepatic transplants in the next 10 to 20 years (133).
Pre-transplant evaluation of patients with NASH or cryptogenic cirrhosis
UNOS recently published that the combination of a high MELD score with severe obesity (BMI>40) decreases survival after a transplant (18). Now many medical centers consider BMI>40 to be an absolute counter indication for hepatic transplants.
Post transplant NAFLD
Survival and management of metabolic syndrome after a transplant
Comparing ages, sexes, MELD scores, and times after transplant among patients who have received liver transplants, patients who had transplants because of NASH compare favorably to other transplant patients. Nevertheless, recipients with NASH who are over the age of 60, have BMIs great than 30 kg/m2 and who had pre-transplant diagnoses of diabetes mellitus and arterial hypertension have a higher first year mortality rate due to cardiovascular complications than do other patients (134).
The components of metabolic syndrome (diabetes, arterial hypertension, dyslipidemia and obesity) are exacerbated by immunosuppression, and are predictors for morbidity and mortality due to cardiovascular causes (129, 135). Corticosteroids cause insulin resistance, fat deposits in the trunk, arterial hypertension, and dyslipidemia. In addition to increased oxidative stress and peroxidation of lipids, calcineurin inhibitors generally cause hyperlipidemia, arterial hypertension and renal damage. For these reasons corticosteroids should be avoided and the use of calcineurin inhibitors should be minimized for these patients. Similarly, since many aspects of this syndrome can be modified, early recognition and treatment of risk factors can have direct impacts on the survival of these patients (18, 136).
De novo NASH
De Novo NAFLD has been described in 20% of hepatic transplant recipients with no prior history of fatty liver disease, while de Novo NASH has been described in 10% of these patients (18). Dumortier et al. (137) demonstrated that post transplant NAFLD is as closely related to the condition of the recipient as it is to the condition of the donor. Post transplant obesity, the use of Tacrolimus, a medical history of diabetes mellitus, hyperlipidemia and/or portal hypertension, a diagnosis of alcoholic cirrhosis as the indication for the hepatic transplant, and steatosis of the allograph are the principal risk factors for the development of de novo NAFLD/NASH following a transplant. Dumortiers study provided the largest cohort of NAFL/NASH patients (421 patients) to be studied to date (137, 138).
References
1. Adler M, Schaffber F. Fatty liver hepatitis and cirrhosis in obese patients. Am J Med 1979; 67: 811-16.
2. Ludwig J, Viaggiano TR, McGill DB, Oh BJ. Non-alcoholic steatohepatitis: Mayo clinic experience with a hitherto unnamed disease. Mayo Clin Proc 1980; 55: 434-8.
3. Erby JR, Silberman C, Lydick E. Prevalence of abnormal serum alanine aminotransferase levels in obese patients and patients with type 2 diabetes. Am J Med 2000; 109: 588-90.
4. Matteoni CA, Younossi ZM, Gramlich T, et al. Non-alcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413-9.
5. Caldwell SH, Hespenheide EE. Subacute liver failure in obese women. Am J Gastroenterol 2002; 97: 2058-62.
6. Shimada M, Hashimoto E, Tania M, et al. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 2002; 37: 154-60.
7. Hashizume H, Sato K, Takagi H, et al. Primary liver cancers with nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 2007; 19: 827-34.
8. Sanyal AJ, Campell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120(5):118392.
9. Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol 2008; 19(3): 295-300.
10. McCullogh AJ. The epidemiology and risk factors of NASH. In Farrel GC, George J, Hall P de la M, et al (eds.) Fatty Liver Disease, NASH and Related Disorders, Malden, MA: Blackwell Publishing 2005. p. 23-37.
11. Wanless IR, Lentz JS. Fatty Liver Hepatitis (Steatohepatitis) and Obesity: An Autopsy Study with Analysis of Risk Factors. Hepatology 1990; 12: 1106-1110.
12. Brunt EM. Histopathology of Non-Alcoholic FattyLiver Disease. Clin Liver Dis 2009; 13: 533-544.
13. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in non-alcoholic fatty liver disease. N Eng J Med 2010; 363: 1341-50.
14. Neuschwander-Tetri BA. Lifestyle modification as the primary treatment of NASH. Clin Liver Dis 2009; 13: 649-665.
15. Ratziu V, Sagi SZ. Pharmacologic therapy of non-alcoholic steatohepatitis. Clin Liver Dis 2009; 13: 667-688.
16. Anjana AP, Rinella ME. Non-alcoholic liver disease: Is bariatric surgery the answer. Clin Liver Dis 2009; 13: 689-710.
17, Musso G, Gambino R, Cassader M, et al. A Meta-Analysis of Randomized Trials for the Treatment of Nonalcoholic Fatty Liver Disease. Hepatology 2010; 52: 79-104.
18. Kymberly DS, Charlton MR. Metabolic syndrome and liver transplantation: A review and guide to management. J Hepatol 2010; 53: 199-206.
19. Hourigan LF, Macdonald GA, Purdie D, et al. Fibrosis in chronic hepatitis C correlates with body mass index and steatosis. Hepatology 1999; 29: 1215-9.
20. Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25: 108-11.
21. Schwimmer JB, Deustch R, Behling C, et al. Obesity, insulin resistance, and other clinicopathological correlations of pediatric non-alcoholic fatty liver disease. J Pediatr 2003; 143: 500-6.
22. Falck-Ytter Y, Younossi ZM, Marchesini G, et al. Clinical features and natural history of non-alcoholic steatosis syndromes. Semin Liver Dis 2001; 21: 17-26.
23. Clark JM, Diehl AM. Defining non-alcoholic liver disease: implications of epidemiological studies. Gastroenterology 2003; 124: 248-50.
24. Seidell JC. Obesity, insulin resistance and diabetes: a world wide epidemic. Br J Nutr 2000; 83(Suppl. 1): 55-8.
25. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Non-alcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 1994; 107: 1103-9.
26. Powell EE, Cooksley WGE, Hanson R, et al. The natural history of non-alcoholic steatohepatitis: a follow-up study of 42 patients for up to 21 years. Hepatology 1990; 11: 74-80.
27. Nomura H, Kashiwagi S, Hayashi J, et al. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn J Med 1988; 27: 142-9.
28. Agarawal SR, Maihotra V, Sakhuja P, Sarin SK. Clinical biochemical and histological profile of non-alcoholic steatohepatitis. Indian J Gastroenterol 2001; 20: 183-6.
29. El-Hassan AY, Ibrahim EM, Al-Mulhim FA, et al. Fatty infiltration of the liver: analysis of prevalence, radiological and clinical features and influence on patient management. Br J Radiol 1992; 65: 774-8.
30. Araujo LM, DeOliveira DA, Nunes DS. Liver and biliary ultrasonography in diabetic and not diabetic obese women. Diabetes Metab 1998; 24: 455-62.
31. Hulcrantz R, Glaumann H, Lindberg J, et al. Liver investigation in 149 asymptomatic patients with moderately elevated activities of serum transaminases. Scand J Gastroenterol 1986; 21: 109-13.
32. Cortez-Pinto H, Camilu ME, Baptista Baptista A, et al. Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr 1999; 18: 353-8.
33. Hasan I, Gani RA, Machmud R, Prevalence and risk factors for non-alcoholic fatty liver in Indonesia. J Gastroenterol Hepatol 2002; 17(Suppl. A): 30.
34. Bellentani S, Saccoccio G, Masatti F, et al. Prevalence and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med 2000; 132: 112-7.
35. Bellentani S, Tiribelli C. The spectrum of liver disease in the general population: lessons from the Dionysos study. J hepatol 2001; 35: 531-7.
36. Omagari KH, Kadokawa Y, Masuda J, et al. Fatty liver in non-alcoholic non-overweight Japanese adults: incidence and clinical characteristics. J Gastroenterol Hepatol 2002; 17: 1089-105.
37. World Health Organization. Obesity: Preventing and Managing the Global Epidemic. Geneva: World Health Organization, 1998.
38. World Health Organization. Definition, diagnosis and classification of diabetes and its complications. I. Diagnosis and classification of diabetes. Geneva: World Health Organization, 1999. p. 20-1.
39. International Diabetes Institute. The Asia-Pacific Perspective: Redefining Obesity and its Treatment. Health Communications Australia, 2000. p. 54.
40. Santos L, Molina EG, Jeffers L, Rajender R, Schiff E. Prevalence of nonalcoholic steatohepalitis among ethnic groups. Gastroenterology 2001; 120(5): A117, Abstract 630.
41. Nonomura A, Mizukami Y, Unoura M. Clinicopathological study of alcoholic-like liver disease in non-alcoholics; non-alcoholic steatohepatitis and fibrosis. Gastroenterol Jpn 1992; 27: 521-8.
42. Propst A, Propst T, Judmaier G, Vogel W. Prognosis in non-alcoholic steatohepatitis [letter]. Gastroenterol 1995; 108: 1607.
43. Skelly MM, James PD, Ryder SD. Findings on liver biopsy to investigate abnormal liver function test in the absence of diagnostic serology. J Hepatol 2001; 35: 195-9.
44. Daniel S, Ben-Menachem T, Vasudevan G, et al. Prospective evaluation of unexplained chronic liver transaminases abnormalities in asymptomatic patients. Am J Gastroenterol 1999; 94: 3010-4.
45. Mathieson NL, Franzen LE, Fryden A, et al. The clinical significance of slightly to moderately increased liver transaminase values in asymptomatic patients. Scand J Gastroenterol 1999; 34: 55-91.
46. Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology 2000; 118: 1117-23.
47. Hilden M, Christoffersen P, Juhl E, et al. Liver histology in "normal" population: examination of 503 consecutive fatal traffic casualties. Scand J Gastroenterol 1977; 12: 593-9.
48. Ground KE. Liver pathology in review. Aviat Space Envirom Med 1982; 53: 14-8.
49. Marcus A, Fisher RA, Jam JM, et al. selection and outcome of living donors for adult right lobe. Transplantation 2000; 69: 2410-5.
50. Marceau P, Biron S, Hould FS, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab 1999; 84: 1513-7.
51. DelGaudio A, Boschi L, DelGaudio GA, et al. Liver damage in obese patients. Obes Surg 2001; 11: 254-7.
52. Lucyckx FA, Desaive C, Tiry A, et al. Liver abnormalities in severe obese subjects: effects of drastic weight loss after gastroplasty. Int J Obes 1988; 22: 222-6.
53. Dixon JR, Bhathal PS, OBrien PE. Non-alcoholic fatty liver disease: predictors of non-alcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001; 121(1): 91-100.
54. Klain J, Fraser D, Goldstein J, et al. Liver histology in the morbidly obese. Hepatology 1989; 10: 873-6.
55. Silverman EM, Sapala JA, Appelman HD, et al. Regression of hepatic steatosis in morbidly obese persons after gastric bypass. Am J Clin Path 1995; 104: 23-31.
56. Ruales FL, Gómez E. Infiltración grasa hepática difusa y su correlación con índice de masa corporal, triglicéridos y transaminasas. Tesis. 2010. http://repository.urosario.edu.co/handle/10336/1965
57. Leonardo A, Bellini M, Tartoni P, et al. The bright liver syndrome: prevalence and determinants of a "bright" liver echo pattern. Ital J Gastroenterol Hepatol 1997; 29: 351-6.
58. Patt CH, Yoo HY, Dibadji K, et al. Prevalence of transaminase abnormalities in asymptomatic health subjects. Dig Dis Sci 2003; 48: 797-801.
59. Clark JM, Brancati FL, Diehl AM. Non-alcoholic fatty liver disease. Gastroenterology 2002; 122: 1649-57.
60. Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the Unites States. Am J Gastroenterol 2003; 98: 960-7.
61. Ruhl C, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology 2003; 124: 71-9.
62. Knobler H, Schattner A, Zhornick T, et al. Fatty liver: an additional and treatable feature of the insulin resistance syndrome. Q J Med 1999; 92: 73-9.
63. Lee RG. Non-alcoholic steatohepatitis: a study of 49 patients. Hum Pathol 1988; 20: 594-8.
64. Rashid M, Roberts E. Non-alcoholic steatohepatitis in children. J Pediatric Gastroenterol Nutr 2000; 30: 48-53.
65. McCullogh AJ, Falck-Ytter Y. Body composition and hepatic steatosis as precursors for fibrotic liver disease. Hepatology 1999; 29: 1328-39.
66. Katsuki A, Sumida T, Murashima S, et al. Serum levels of tumor necrosis factor a are increased in obese patients with non-insulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1998; 83: 859-62.
67. Shuldiner AR, Yang R, Gong DW. Resistin, Obesity and insulin resistance: the emerging role of the adipocyte as an endocrine organ. N eng J Med 2001; 345: 1345-6.
68. Lonquist F, Arner P, Nordfors L, et al. Over expression of the obese (OB) gene in adipose tissue of human obese subjects. Nat Med 1995; 1: 950-3.
69. Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab 2000; 11: 351-6.
70. Bergh AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metabol 2002; 13: 84-9.
71. Kral JG, Schaffner F, Pierson RN et al. Body fat topography as an independent predictor of fatty liver. Metabolism 1993; 42: 548-51.
72. Peiris AN, Mueller RA, Smith GA et al. Splachnic insulin metabolism in obesity: influence of body fat distribution. J Clin Invest 1986; 78: 1648-57.
73. Younossi ZM, Gramlich T, Matteoni CA, et al. Non-alcoholic fatty liver disease in patients with type II diabetes. Clin Gastroenterol Hepatol 2004; 2: 262-5.
74. Angulo P, Keach JC, Batts KP, et al. Independent predictors of liver fibrosis in patients with non-alcoholic steatohepatitis. Hepatology 1999; 30: 1356-62.
75. George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in non-alcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998; 114: 311-8.
76. Saadeh S, Younossi ZM, Remer EM, et al. The utility of radiological imaging in non-alcoholic fatty liver disease. Gastroenterology 2002; 123: 745-50.
77. Gaede P, Vedel P, Larsen N et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Eng J Med 2003; 348: 383-93.
78. Sasaki A, Horiuchi N, Hasegawa K. et al. Mortality and causes of death in type 2 diabetic patients. Diabetes Res Clin Pract 1989; 7: 33-40.
79. Ayata G, Gordon FD, Lewis WD. et al. Cryptogenic cirrhosis: clinicopathologic findings at and after liver transplantation. Hum Pathol 2002; 33: 1098-104.
80. Nosaldini R, Avogaro A, Mollo F, et al. Carbohidrate and lipid metabolism in cirrhosis: evidence that hepatic uptake of gluconeogenic precursors and of free fatty acids depends on effective hepatic blood flow. J Clin Endocrinol Metab 1984; 58: 1125-32.
81. Wanless IR, Wong F, Blendis LM, et al. Hepatic and portal vein thrombosis in cirrhosis: possible role in the development of parenchymal extinction and portal hypertension. Hepatology 1995; 21: 1238-47.
82. Ratziu V, Bonyhay L, Di Martino V, et al. Survival, liver failure and hepatocellular carcinoma in obesity-related cryptogenic cirrhosis. Hepatology 2002; 35: 1485-93.
83. Bugianasi E, Leone N, Vanni E, et al. Expanding the natural history of non-alcoholic steatohepatitis from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002; 123: 134-40.
84. Petta S, Muratore C, A Craxi. Non-alcoholic fatty liver disease pathogenesis. Digestive and Liver Disease 2009; 4: 615-622.
85. Marchesini G, Bugianesi E, Forlani G, Cerreli F, Lenzi M, et al. Non-alcoholic fatty liver, steatohepatitis and the Metabolic Syndrome. Hepatology 2003; 37: 917-23.
86. Day CP. From fat to inflammation. Gastroenterology 2006; 130: 207-210.
87. De Alwis NM, Day CP. Genetic of alcohol liver disease and Non-alcoholic fatty liver disease. Sem Liver Dis 2007; 27: 44-54.
88. Córdova Pluma VH. Hígado graso no alcohólico: un encuadre didáctico para un problema latente. Segunda parte. Med Int Mex 2009; 25(2): 129-53
89. Gastaldelli A, Natali A, Vettor R, Corradini. Insulina resistance, adipose depots and gut: interactions and pathological implications. Digestive and Liver Disease 2010; 42: 310-319
90. Vanni E, Bugianesi E, Kotronen A, et al. From the metabolic to NAFLD o vice versa? Digestive and liver Disease 2010; 42: 320-330.
91. Bugianesi E, McCullough AJ, Marchenisi G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 2005; 42: 987-1000.
92. Angulo P. Non-alcoholic fatty liver. N Engl J Med 2002; 346: 1221.
93. Adams LA, Feldstein A. Nonalcoholic Steatohepatitis: Risk factors and Diagnosis. Expert Rev Gastroentrol Hepatol 2010; 4(5): 623-635.
94. Feldstedin AE, Wieckowska A, López A, et al. Cytokeratins -18 fragment for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 2009; 50: 1072-1078.
95. Chavez-Tapia NC, Uribe M, Ponciano-Rodriguez G, Medina-Satillan R, Mendez-Sanchez N. New Insights into the pathophysiology of non-alcoholic fatty liver. Annals of Hepatology 2009; 8: S9-S17.
96. Wigg AJ, Roberts-Thoson IC, Dymock ER, et al. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia and tumor necrosis factor alpha in the pathogenesis of nonalcoholic steatohepatitis. Gut 2001; 48: 206.
97. Tilg H, Moschen A. Evolution of inflammation in Non alcoholic Fatty Liver Disease: The multiple Parallel Hits Hypothesis. Hepatology 2010; 52: 1836-1846.
98. Harrison SA, Diehl AM. Fat and the liver: a molecular overview. Seminar Gastrointest Dis 2002; 13: 3-16.
99. Harrison SA, Hayashi P. Clinical factors associated with fibrosis in 102 patientes with non-alcoholic steatohepatitis [Abstract]. Hepatology 2002; 36: 412A.
100. Harrison SA, Kadakia S, Lang KA, et al. Non-alcoholic steatohepatitis: What we know in the new millenium. Am J Gastroenterol 2002; 97: 2714-24.
101. Kumar KS, Malet PF. Non-alcoholic steatohepatitis. Mayo Clinic Proc 2000; 75: 733-9.
102. Xiaozhou Ma, Halalkere NS, Kambadakone A, Kenudson MM, Hahn P, Sahani D. Imaging-based quantification of hepatic fat: methods and clinical applications. RadioGraphics 2009, 29: 1253-1277.
103. Kadama Y, Ng CS, Wu TT. Comparison of CT methods for determining the fat content of the liver. Am J Roentgenol 2007;188: 1307-1312.
104. Cowin GJ, Jonsson JR, Bauer JD. Magnetic resonance imaging and spectroscopy for monitoring liver steatosis. J Magn Reson Imaging 2008; 28: 937-945.
105. Estep JM, Bererdinc A, Younossi Z. Non-Invasive Diagnosatic Test forNon-Alcoholic Fatty Liver Disease. 2010, Current Molecular Medicine 2010; 10: 166-172.
106. Brunt EM. Non-alcoholic fatty liver disease. In Burt AD, Portmann BC, Ferrel LD (eds): Mc Sweens Pathology of the Liver, Elsevier Publishing 2007. p. 367-291.
107. Burt AD, Mutton A, Day CP, et al. Diagnosis and interpretation of steatosis and stetatohepatitis. Semin Diagn Pathol 1998; 15: 246-58.
108. Kleiner De, Brunt EM, Van Natta M, et al. Non-alcoholic steatohepatitis Clinical Research Network: Design and validation of a histological scoring system for non-alcoholic fatty liver disease. Hepatology 2005; 41: 1313-1321.
109. Brunt E. Pathology of Fatty Liver Disease. Modern Pathology 2007; 20: S40-S48.
110. Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic Steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003; 37: 1202-1219.
111. Law K, Brunt EM. Non-alcoholic fatty liver disease. Clin Liver Dis 2010; 14: 591-604.
112. Ratziu V, Bellenteni S, Cortez-Pinto, Day C, Marchesini G. A position statement on NAFLD|NASH based on the EASL 2009 special conference. J Hepatol 2010; 53: 372-384.
113. Rafiq N, Younossi Z. Non-alcoholic fatty liver Disease: a practical approach to evaluation and management. Clin Liver Dis 2009; 13: 249-266.
114. Ratziu V, Zelber-Sagi S. Pharmacologic therapy of Non alcoholic Steatohepatitis 2009; 13: 667-688.
115. Suzuky A. Lindor K, St Saver, et al. effect of changes on body weight and lifestyle in Non-alcoholic fatty liver. J Hepatol 2005; 43: 1060-1066.
116. Musso G, Gambino R, Michieli F, et al. Dietary habits and their relation to insulin resistance and postprandial lipemia in non-alcoholic steatohepatitis. Hepatology 2003; 37: 909-916.
117. Sacks FM, Bray GA, Carey V, et al. Comparison of weight loss diets with differents compositions of fat, protein and carbohydrates. N Engl J Med 2009; 360: 859-873.
118. Ouyan X, Cirillo P, Sautin Y, Mccall S, Bruchete JL, et al. Fructose consumption as a risk factor for non alcoholic fatty liver disease. J Hepatol 2008; 48: 993-999.
119. Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decrease prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47: 1947-1954.
120. Frith J, Day CP, Robinson L, et al. Potential strategies to improve uptake of exercise interventions in nonalcoholic fatty liver disease. J Hepatol 2010; 52: 112-116.
121. Krasnoff JB, Painter PL, Wallace JP. Health related fitness and physical activity in patients with nonalcoholic liver disease. Hepatology 2008; 47: 1158-1166.
122. Klein S, Mittendorfer B, Eagon JC, et al. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic liver disease. Gastroenterology 2006; 130: 1564-1772.
123. Mummadi RR, Kasturi KS, et al. effect bariatric surgery on nonalcoholic liver disease: systematic review and meta-analysis. Clin gastroenterol hepatol 2008; 6: 1396-402.
124. Chávez-Tapia NC, Téllez-Ávila FL, et al. Bariatric surgery for nonalcoholic steatohepatitis in obese patients. Cochrane database Syst rev 2010:1 CD007340
125. Loria P, Adinolfi LE, Bellentani S, Bugianesi E, et al. Practice guidelines for the diagnosis and manangment of nonalcoholic fatty liver disease. Digestive and Liver Disease 2010; 42: 272-282.
126. Burke A, Lucey MR. NAFLD, NASH and orthotopic liver transplantation. In Farrel GC, George J, Hall P de la M et al (eds) Fatty Liver Disease: NASH and Related Disorders, Malden, MA: Blackwell Publishing 2005. p. 208-217.
127. Adam R, Reynes M, Johann M, et al. The outcome of steatotic grafts in liver transplantation. Transplant Proc 1991; 23(1 Pt 2): 1538-1540.
128. Tevar AD, Clarke C, Wang J, Rudich SM, Woodle ES, Lentsch AB, et al. Clinical review of nonalcoholic steatohepatitis in liver surgery and transplantation. J Am Coll Surg 2010; 210(4): 515-526.
129. Yalamanchili K, Saadeh S, Klintmalm GB, Jennings LW, Davis GL. Nonalcoholic fatty liver disease after liver transplantation for cryptogenic cirrhosis or nonalcoholic fatty liver disease. Liver Transpl 2010; 16: 431-439.
130. Nayak NC, Vasdev N, Saigal S, Soin AS. End-stage nonalcoholic fatty liver disease: evaluation of pathomorphologic features and relationship to cryptogenic cirrhosis from study of explant livers in a living donor liver transplant program. Human Pathology 2010; 41: 425-430.
131. Malik SM, Gupte PA, deVera ME, Ahmad J. Liver transplantation in patients with nonalcoholic steatohepatitis-related hepatocellular carcinoma. Clinical gastroenterology and hepatology 2009; 7: 800-806.
132. Argo CK, Caldwell SH. Epidemiology and natural history of nonalcoholic steatohepatitis. Clin Liver Dis 2009; 13: 511-531.
133. Charlton M. Obesity, Hyperlipidemia and Metabolic Syndrome. Liver Transplantation 2009; 15(11): S83-S89.
134. Malik SM, deVera ME, Fontes P, Shaikh O, Ahmad J. Outcome after liver transplantation for NASH cirrhosis. American Journal of Transplantation 2009; 9: 782-793.
135. Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15: 1662-1670.
136. Koehler E, Watt K, Charlton M. Fatty Liver and Liver Transplantation. Clin Liver Dis 2009; 13: 621-630.
137. Dumortier J, Giostra E, Belbouab S, Morard I, Guillaud O, Spahr L, et al. Non-alcoholic fatty liver disease in liver transplant recipients: another story of "seed and soil". Am J Gastroenterol 2010; 105: 613-620.
138. Ong J, Younossi ZM. Non-alcoholic fatty liver disease after liver transplantation: a case of nurture and nature. Editorial. Am J Gastroenterol 2010; 105: 621-623.
1. Adler M, Schaffber F. Fatty liver hepatitis and cirrhosis in obese patients. Am J Med 1979; 67: 811-16. [ Links ]
2. Ludwig J, Viaggiano TR, McGill DB, Oh BJ. Non-alcoholic steatohepatitis: Mayo clinic experience with a hitherto unnamed disease. Mayo Clin Proc 1980; 55: 434-8. [ Links ]
3. Erby JR, Silberman C, Lydick E. Prevalence of abnormal serum alanine aminotransferase levels in obese patients and patients with type 2 diabetes. Am J Med 2000; 109: 588-90. [ Links ]
4. Matteoni CA, Younossi ZM, Gramlich T, et al. Non-alcoholic fatty liver disease: a spectrum of clinical and pathological severity. Gastroenterology 1999; 116: 1413-9. [ Links ]
5. Caldwell SH, Hespenheide EE. Subacute liver failure in obese women. Am J Gastroenterol 2002; 97: 2058-62. [ Links ]
6. Shimada M, Hashimoto E, Tania M, et al. Hepatocellular carcinoma in patients with non-alcoholic steatohepatitis. J Hepatol 2002; 37: 154-60. [ Links ]
7. Hashizume H, Sato K, Takagi H, et al. Primary liver cancers with nonalcoholic steatohepatitis. Eur J Gastroenterol Hepatol 2007; 19: 827-34. [ Links ]
8. Sanyal AJ, Campell-Sargent C, Mirshahi F, et al. Nonalcoholic steatohepatitis: association of insulin resistance and mitochondrial abnormalities. Gastroenterology 2001; 120(5):1183–92. [ Links ]
9. Choi SS, Diehl AM. Hepatic triglyceride synthesis and nonalcoholic fatty liver disease. Curr Opin Lipidol 2008; 19(3): 295-300. [ Links ]
10. McCullogh AJ. The epidemiology and risk factors of NASH. In Farrel GC, George J, Hall P de la M, et al (eds.) Fatty Liver Disease, NASH and Related Disorders, Malden, MA: Blackwell Publishing 2005. p. 23-37. [ Links ]
11. Wanless IR, Lentz JS. Fatty Liver Hepatitis (Steatohepatitis) and Obesity: An Autopsy Study with Analysis of Risk Factors. Hepatology 1990; 12: 1106-1110. [ Links ]
12. Brunt EM. Histopathology of Non-Alcoholic FattyLiver Disease. Clin Liver Dis 2009; 13: 533-544. [ Links ]
13. Targher G, Day CP, Bonora E. Risk of cardiovascular disease in non-alcoholic fatty liver disease. N Eng J Med 2010; 363: 1341-50. [ Links ]
14. Neuschwander-Tetri BA. Lifestyle modification as the primary treatment of NASH. Clin Liver Dis 2009; 13: 649-665. [ Links ]
15. Ratziu V, Sagi SZ. Pharmacologic therapy of non-alcoholic steatohepatitis. Clin Liver Dis 2009; 13: 667-688. [ Links ]
16. Anjana AP, Rinella ME. Non-alcoholic liver disease: Is bariatric surgery the answer. Clin Liver Dis 2009; 13: 689-710. [ Links ]
17. Musso G, Gambino R, Cassader M, et al. A Meta-Analysis of Randomized Trials for the Treatment of Nonalcoholic Fatty Liver Disease. Hepatology 2010; 52: 79-104. [ Links ]
18. Kymberly DS, Charlton MR. Metabolic syndrome and liver transplantation: A review and guide to management. J Hepatol 2010; 53: 199-206. [ Links ]
19. Hourigan LF, Macdonald GA, Purdie D, et al. Fibrosis in chronic hepatitis C correlates with body mass index and steatosis. Hepatology 1999; 29: 1215-9. [ Links ]
20. Naveau S, Giraud V, Borotto E, et al. Excess weight risk factor for alcoholic liver disease. Hepatology 1997; 25: 108-11. [ Links ]
21. Schwimmer JB, Deustch R, Behling C, et al. Obesity, insulin resistance, and other clinicopathological correlations of pediatric non-alcoholic fatty liver disease. J Pediatr 2003; 143: 500-6. [ Links ]
22. Falck-Ytter Y, Younossi ZM, Marchesini G, et al. Clinical features and natural history of non-alcoholic steatosis syndromes. Semin Liver Dis 2001; 21: 17-26. [ Links ]
23. Clark JM, Diehl AM. Defining non-alcoholic liver disease: implications of epidemiological studies. Gastroenterology 2003; 124: 248-50. [ Links ]
24. Seidell JC. Obesity, insulin resistance and diabetes: a world wide epidemic. Br J Nutr 2000; 83(Suppl. 1): 55-8. [ Links ]
25. Bacon BR, Farahvash MJ, Janney CG, Neuschwander-Tetri BA. Non-alcoholic steatohepatitis: an expanded clinical entity. Gastroenterology 1994; 107: 1103-9. [ Links ]
26. Powell EE, Cooksley WGE, Hanson R, et al. The natural history of non-alcoholic steatohepatitis: a follow-up study of 42 patients for up to 21 years. Hepatology 1990; 11: 74-80. [ Links ]
27. Nomura H, Kashiwagi S, Hayashi J, et al. Prevalence of fatty liver in a general population of Okinawa, Japan. Jpn J Med 1988; 27: 142-9. [ Links ]
28. Agarawal SR, Maihotra V, Sakhuja P, Sarin SK. Clinical biochemical and histological profile of non-alcoholic steatohepatitis. Indian J Gastroenterol 2001; 20: 183-6. [ Links ]
29. El-Hassan AY, Ibrahim EM, Al-Mulhim FA, et al. Fatty infiltration of the liver: analysis of prevalence, radiological and clinical features and influence on patient management. Br J Radiol 1992; 65: 774-8. [ Links ]
30. Araujo LM, DeOliveira DA, Nunes DS. Liver and biliary ultrasonography in diabetic and not diabetic obese women. Diabetes Metab 1998; 24: 455-62. [ Links ]
31. Hulcrantz R, Glaumann H, Lindberg J, et al. Liver investigation in 149 asymptomatic patients with moderately elevated activities of serum transaminases. Scand J Gastroenterol 1986; 21: 109-13. [ Links ]
32. Cortez-Pinto H, Camilu ME, Baptista Baptista A, et al. Non-alcoholic fatty liver: another feature of the metabolic syndrome? Clin Nutr 1999; 18: 353-8. [ Links ]
33. Hasan I, Gani RA, Machmud R, Prevalence and risk factors for non-alcoholic fatty liver in Indonesia. J Gastroenterol Hepatol 2002; 17(Suppl. A): 30. [ Links ]
34. Bellentani S, Saccoccio G, Masatti F, et al. Prevalence and risk factors for hepatic steatosis in Northern Italy. Ann Intern Med 2000; 132: 112-7. [ Links ]
35. Bellentani S, Tiribelli C. The spectrum of liver disease in the general population: lessons from the Dionysos study. J hepatol 2001; 35: 531-7. [ Links ]
36. Omagari KH, Kadokawa Y, Masuda J, et al. Fatty liver in non-alcoholic non-overweight Japanese adults: incidence and clinical characteristics. J Gastroenterol Hepatol 2002; 17: 1089-105. [ Links ]
37. World Health Organization. Obesity: Preventing and Managing the Global Epidemic. Geneva: World Health Organization, 1998. [ Links ]
38. World Health Organization. Definition, diagnosis and classification of diabetes and its complications. I. Diagnosis and classification of diabetes. Geneva: World Health Organization, 1999. p. 20-1. [ Links ]
39. International Diabetes Institute. The Asia-Pacific Perspective: Redefining Obesity and its Treatment. Health Communications Australia, 2000. p. 54. [ Links ]
40. Santos L, Molina EG, Jeffers L, Rajender R, Schiff E. Prevalence of nonalcoholic steatohepalitis among ethnic groups. Gastroenterology 2001; 120(5): A117, Abstract 630. [ Links ]
41. Nonomura A, Mizukami Y, Unoura M. Clinicopathological study of alcoholic-like liver disease in non-alcoholics; non-alcoholic steatohepatitis and fibrosis. Gastroenterol Jpn 1992; 27: 521-8. [ Links ]
42. Propst A, Propst T, Judmaier G, Vogel W. Prognosis in non-alcoholic steatohepatitis [letter]. Gastroenterol 1995; 108: 1607. [ Links ]
43. Skelly MM, James PD, Ryder SD. Findings on liver biopsy to investigate abnormal liver function test in the absence of diagnostic serology. J Hepatol 2001; 35: 195-9. [ Links ]
44. Daniel S, Ben-Menachem T, Vasudevan G, et al. Prospective evaluation of unexplained chronic liver transaminases abnormalities in asymptomatic patients. Am J Gastroenterol 1999; 94: 3010-4. [ Links ]
45 Mathieson NL, Franzen LE, Fryden A, et al. The clinical significance of slightly to moderately increased liver transaminase values in asymptomatic patients. Scand J Gastroenterol 1999; 34: 55-91. [ Links ]
46. Ratziu V, Giral P, Charlotte F, et al. Liver fibrosis in overweight patients. Gastroenterology 2000; 118: 1117-23. [ Links ]
47. Hilden M, Christoffersen P, Juhl E, et al. Liver histology in "normal" population: examination of 503 consecutive fatal traffic casualties. Scand J Gastroenterol 1977; 12: 593-9. [ Links ]
48. Ground KE. Liver pathology in review. Aviat Space Envirom Med 1982; 53: 14-8. [ Links ]
49. Marcus A, Fisher RA, Jam JM, et al. selection and outcome of living donors for adult right lobe. Transplantation 2000; 69: 2410-5. [ Links ]
50. Marceau P, Biron S, Hould FS, et al. Liver pathology and the metabolic syndrome X in severe obesity. J Clin Endocrinol Metab 1999; 84: 1513-7. [ Links ]
51. DelGaudio A, Boschi L, DelGaudio GA, et al. Liver damage in obese patients. Obes Surg 2001; 11: 254-7. [ Links ]
52. Lucyckx FA, Desaive C, Tiry A, et al. Liver abnormalities in severe obese subjects: effects of drastic weight loss after gastroplasty. Int J Obes 1988; 22: 222-6. [ Links ]
53. Dixon JR, Bhathal PS, OBrien PE. Non-alcoholic fatty liver disease: predictors of non-alcoholic steatohepatitis and liver fibrosis in the severely obese. Gastroenterology 2001; 121(1): 91-100. [ Links ]
54. Klain J, Fraser D, Goldstein J, et al. Liver histology in the morbidly obese. Hepatology 1989; 10: 873-6. [ Links ]
55. Silverman EM, Sapala JA, Appelman HD, et al. Regression of hepatic steatosis in morbidly obese persons after gastric bypass. Am J Clin Path 1995; 104: 23-31. [ Links ]
56. Ruales FL, Gómez E. Infiltración grasa hepática difusa y su correlación con índice de masa corporal, triglicéridos y transaminasas. Tesis. 2010. http://repository.urosario.edu.co/handle/10336/1965 [ Links ]
57. Leonardo A, Bellini M, Tartoni P, et al. The bright liver syndrome: prevalence and determinants of a "bright" liver echo pattern. Ital J Gastroenterol Hepatol 1997; 29: 351-6. [ Links ]
58. Patt CH, Yoo HY, Dibadji K, et al. Prevalence of transaminase abnormalities in asymptomatic health subjects. Dig Dis Sci 2003; 48: 797-801. [ Links ]
59. Clark JM, Brancati FL, Diehl AM. Non-alcoholic fatty liver disease. Gastroenterology 2002; 122: 1649-57. [ Links ]
60. Clark JM, Brancati FL, Diehl AM. The prevalence and etiology of elevated aminotransferase levels in the Unites States. Am J Gastroenterol 2003; 98: 960-7. [ Links ]
61. Ruhl C, Everhart JE. Determinants of the association of overweight with elevated serum alanine aminotransferase activity in the United States. Gastroenterology 2003; 124: 71-9. [ Links ]
62. Knobler H, Schattner A, Zhornick T, et al. Fatty liver: an additional and treatable feature of the insulin resistance syndrome. Q J Med 1999; 92: 73-9. [ Links ]
63. Lee RG. Non-alcoholic steatohepatitis: a study of 49 patients. Hum Pathol 1988; 20: 594-8. [ Links ]
64. Rashid M, Roberts E. Non-alcoholic steatohepatitis in children. J Pediatric Gastroenterol Nutr 2000; 30: 48-53. [ Links ]
65. McCullogh AJ, Falck-Ytter Y. Body composition and hepatic steatosis as precursors for fibrotic liver disease. Hepatology 1999; 29: 1328-39. [ Links ]
66. Katsuki A, Sumida T, Murashima S, et al. Serum levels of tumor necrosis factor a are increased in obese patients with non-insulin-dependent diabetes mellitus. J Clin Endocrinol Metab 1998; 83: 859-62. [ Links ]
67. Shuldiner AR, Yang R, Gong DW. Resistin, Obesity and insulin resistance: the emerging role of the adipocyte as an endocrine organ. N eng J Med 2001; 345: 1345-6. [ Links ]
68. Lonquist F, Arner P, Nordfors L, et al. Over expression of the obese (OB) gene in adipose tissue of human obese subjects. Nat Med 1995; 1: 950-3. [ Links ]
69. Bergman RN, Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol Metab 2000; 11: 351-6. [ Links ]
70. Bergh AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metabol 2002; 13: 84-9. [ Links ]
71. Kral JG, Schaffner F, Pierson RN et al. Body fat topography as an independent predictor of fatty liver. Metabolism 1993; 42: 548-51. [ Links ]
72. Peiris AN, Mueller RA, Smith GA et al. Splachnic insulin metabolism in obesity: influence of body fat distribution. J Clin Invest 1986; 78: 1648-57. [ Links ]
73. Younossi ZM, Gramlich T, Matteoni CA, et al. Non-alcoholic fatty liver disease in patients with type II diabetes. Clin Gastroenterol Hepatol 2004; 2: 262-5. [ Links ]
74. Angulo P, Keach JC, Batts KP, et al. Independent predictors of liver fibrosis in patients with non-alcoholic steatohepatitis. Hepatology 1999; 30: 1356-62. [ Links ]
75. George DK, Goldwurm S, MacDonald GA, et al. Increased hepatic iron concentration in non-alcoholic steatohepatitis is associated with increased fibrosis. Gastroenterology 1998; 114: 311-8. [ Links ]
76. Saadeh S, Younossi ZM, Remer EM, et al. The utility of radiological imaging in non-alcoholic fatty liver disease. Gastroenterology 2002; 123: 745-50. [ Links ]
77. Gaede P, Vedel P, Larsen N et al. Multifactorial intervention and cardiovascular disease in patients with type 2 diabetes. N Eng J Med 2003; 348: 383-93. [ Links ]
78. Sasaki A, Horiuchi N, Hasegawa K. et al. Mortality and causes of death in type 2 diabetic patients. Diabetes Res Clin Pract 1989; 7: 33-40. [ Links ]
79. Ayata G, Gordon FD, Lewis WD. et al. Cryptogenic cirrhosis: clinicopathologic findings at and after liver transplantation. Hum Pathol 2002; 33: 1098-104. [ Links ]
80. Nosaldini R, Avogaro A, Mollo F, et al. Carbohidrate and lipid metabolism in cirrhosis: evidence that hepatic uptake of gluconeogenic precursors and of free fatty acids depends on effective hepatic blood flow. J Clin Endocrinol Metab 1984; 58: 1125-32. [ Links ]
81. Wanless IR, Wong F, Blendis LM, et al. Hepatic and portal vein thrombosis in cirrhosis: possible role in the development of parenchymal extinction and portal hypertension. Hepatology 1995; 21: 1238-47. [ Links ]
82. Ratziu V, Bonyhay L, Di Martino V, et al. Survival, liver failure and hepatocellular carcinoma in obesity-related cryptogenic cirrhosis. Hepatology 2002; 35: 1485-93. [ Links ]
83. Bugianasi E, Leone N, Vanni E, et al. Expanding the natural history of non-alcoholic steatohepatitis from cryptogenic cirrhosis to hepatocellular carcinoma. Gastroenterology 2002; 123: 134-40. [ Links ]
84. Petta S, Muratore C, A Craxi. Non-alcoholic fatty liver disease pathogenesis. Digestive and Liver Disease 2009; 4: 615-622. [ Links ]
85. Marchesini G, Bugianesi E, Forlani G, Cerreli F, Lenzi M, et al. Non-alcoholic fatty liver, steatohepatitis and the Metabolic Syndrome. Hepatology 2003; 37: 917-23. [ Links ]
86. Day CP. From fat to inflammation. Gastroenterology 2006; 130: 207-210. [ Links ]
87. De Alwis NM, Day CP. Genetic of alcohol liver disease and Non-alcoholic fatty liver disease. Sem Liver Dis 2007; 27: 44-54. [ Links ]
88. Córdova Pluma VH. Hígado graso no alcohólico: un encuadre didáctico para un problema latente. Segunda parte. Med Int Mex 2009; 25(2): 129-53 [ Links ]
89. Gastaldelli A, Natali A, Vettor R, Corradini. Insulina resistance, adipose depots and gut: interactions and pathological implications. Digestive and Liver Disease 2010; 42: 310-319 [ Links ]
90. Vanni E, Bugianesi E, Kotronen A, et al. From the metabolic to NAFLD o vice versa? Digestive and liver Disease 2010; 42: 320-330. [ Links ]
91. Bugianesi E, McCullough AJ, Marchenisi G. Insulin resistance: a metabolic pathway to chronic liver disease. Hepatology 2005; 42: 987-1000. [ Links ]
92. Angulo P. Non-alcoholic fatty liver. N Engl J Med 2002; 346: 1221. [ Links ]
93. Adams LA, Feldstein A. Nonalcoholic Steatohepatitis: Risk factors and Diagnosis. Expert Rev Gastroentrol Hepatol 2010; 4(5): 623-635. [ Links ]
94. Feldstedin AE, Wieckowska A, López A, et al. Cytokeratins -18 fragment for nonalcoholic steatohepatitis: a multicenter validation study. Hepatology 2009; 50: 1072-1078. [ Links ]
95. Chavez-Tapia NC, Uribe M, Ponciano-Rodriguez G, Medina-Satillan R, Mendez-Sanchez N. New Insights into the pathophysiology of non-alcoholic fatty liver. Annals of Hepatology 2009; 8: S9-S17. [ Links ]
96. Wigg AJ, Roberts-Thoson IC, Dymock ER, et al. The role of small intestinal bacterial overgrowth, intestinal permeability, endotoxaemia and tumor necrosis factor alpha in the pathogenesis of nonalcoholic steatohepatitis. Gut 2001; 48: 206. [ Links ]
97. Tilg H, Moschen A. Evolution of inflammation in Non alcoholic Fatty Liver Disease: The multiple Parallel Hits Hypothesis. Hepatology 2010; 52: 1836-1846. [ Links ]
98. Harrison SA, Diehl AM. Fat and the liver: a molecular overview. Seminar Gastrointest Dis 2002; 13: 3-16. [ Links ]
99. Harrison SA, Hayashi P. Clinical factors associated with fibrosis in 102 patientes with non-alcoholic steatohepatitis [Abstract]. Hepatology 2002; 36: 412A. [ Links ]
100. Harrison SA, Kadakia S, Lang KA, et al. Non-alcoholic steatohepatitis: What we know in the new millenium. Am J Gastroenterol 2002; 97: 2714-24. [ Links ]
101. Kumar KS, Malet PF. Non-alcoholic steatohepatitis. Mayo Clinic Proc 2000; 75: 733-9. [ Links ]
102. Xiaozhou Ma, Halalkere NS, Kambadakone A, Kenudson MM, Hahn P, Sahani D. Imaging-based quantification of hepatic fat: methods and clinical applications. RadioGraphics 2009, 29: 1253-1277. [ Links ]
103. Kadama Y, Ng CS, Wu TT. Comparison of CT methods for determining the fat content of the liver. Am J Roentgenol 2007;188: 1307-1312. [ Links ]
104. Cowin GJ, Jonsson JR, Bauer JD. Magnetic resonance imaging and spectroscopy for monitoring liver steatosis. J Magn Reson Imaging 2008; 28: 937-945. [ Links ]
105. Estep JM, Bererdinc A, Younossi Z. Non-Invasive Diagnosatic Test forNon-Alcoholic Fatty Liver Disease. 2010, Current Molecular Medicine 2010; 10: 166-172. [ Links ]
106. Brunt EM. Non-alcoholic fatty liver disease. In Burt AD, Portmann BC, Ferrel LD (eds): Mc Sweens Pathology of the Liver, Elsevier Publishing 2007. p. 367-291. [ Links ]
107. Burt AD, Mutton A, Day CP, et al. Diagnosis and interpretation of steatosis and stetatohepatitis. Semin Diagn Pathol 1998; 15: 246-58. [ Links ]
108. Kleiner De, Brunt EM, Van Natta M, et al. Non-alcoholic steatohepatitis Clinical Research Network: Design and validation of a histological scoring system for non-alcoholic fatty liver disease. Hepatology 2005; 41: 1313-1321. [ Links ]
109. Brunt E. Pathology of Fatty Liver Disease. Modern Pathology 2007; 20: S40-S48. [ Links ]
110. Neuschwander-Tetri BA, Caldwell SH. Nonalcoholic Steatohepatitis: Summary of an AASLD Single Topic Conference. Hepatology 2003; 37: 1202-1219. [ Links ]
111. Law K, Brunt EM. Non-alcoholic fatty liver disease. Clin Liver Dis 2010; 14: 591-604. [ Links ]
112. Ratziu V, Bellenteni S, Cortez-Pinto, Day C, Marchesini G. A position statement on NAFLD|NASH based on the EASL 2009 special conference. J Hepatol 2010; 53: 372-384. [ Links ]
113. Rafiq N, Younossi Z. Non-alcoholic fatty liver Disease: a practical approach to evaluation and management. Clin Liver Dis 2009; 13: 249-266. [ Links ]
114. Ratziu V, Zelber-Sagi S. Pharmacologic therapy of Non alcoholic Steatohepatitis 2009; 13: 667-688. [ Links ]
115. Suzuky A. Lindor K, St Saver, et al. effect of changes on body weight and lifestyle in Non-alcoholic fatty liver. J Hepatol 2005; 43: 1060-1066. [ Links ]
116. Musso G, Gambino R, Michieli F, et al. Dietary habits and their relation to insulin resistance and postprandial lipemia in non-alcoholic steatohepatitis. Hepatology 2003; 37: 909-916. [ Links ]
117. Sacks FM, Bray GA, Carey V, et al. Comparison of weight –loss diets with differents compositions of fat, protein and carbohydrates. N Engl J Med 2009; 360: 859-873. [ Links ]
118. Ouyan X, Cirillo P, Sautin Y, Mccall S, Bruchete JL, et al. Fructose consumption as a risk factor for non –alcoholic fatty liver disease. J Hepatol 2008; 48: 993-999. [ Links ]
119. Dunn W, Xu R, Schwimmer JB. Modest wine drinking and decrease prevalence of suspected nonalcoholic fatty liver disease. Hepatology 2008; 47: 1947-1954. [ Links ]
120. Frith J, Day CP, Robinson L, et al. Potential strategies to improve uptake of exercise interventions in nonalcoholic fatty liver disease. J Hepatol 2010; 52: 112-116. [ Links ]
121. Krasnoff JB, Painter PL, Wallace JP. Health related fitness and physical activity in patients with nonalcoholic liver disease. Hepatology 2008; 47: 1158-1166. [ Links ]
122. Klein S, Mittendorfer B, Eagon JC, et al. Gastric bypass surgery improves metabolic and hepatic abnormalities associated with nonalcoholic liver disease. Gastroenterology 2006; 130: 1564-1772. [ Links ]
123. Mummadi RR, Kasturi KS, et al. effect bariatric surgery on nonalcoholic liver disease: systematic review and meta-analysis. Clin gastroenterol hepatol 2008; 6: 1396-402. [ Links ]
124. Chávez-Tapia NC, Téllez-Ávila FL, et al. Bariatric surgery for nonalcoholic steatohepatitis in obese patients. Cochrane database Syst rev 2010:1 CD007340 [ Links ]
125. Loria P, Adinolfi LE, Bellentani S, Bugianesi E, et al. Practice guidelines for the diagnosis and manangment of nonalcoholic fatty liver disease. Digestive and Liver Disease 2010; 42: 272-282. [ Links ]
126. Burke A, Lucey MR. NAFLD, NASH and orthotopic liver transplantation. In Farrel GC, George J, Hall P de la M et al (eds) Fatty Liver Disease: NASH and Related Disorders, Malden, MA: Blackwell Publishing 2005. p. 208-217. [ Links ]
127. Adam R, Reynes M, Johann M, et al. The outcome of steatotic grafts in liver transplantation. Transplant Proc 1991; 23(1 Pt 2): 1538-1540. [ Links ]
128. Tevar AD, Clarke C, Wang J, Rudich SM, Woodle ES, Lentsch AB, et al. Clinical review of nonalcoholic steatohepatitis in liver surgery and transplantation. J Am Coll Surg 2010; 210(4): 515-526. [ Links ]
129. Yalamanchili K, Saadeh S, Klintmalm GB, Jennings LW, Davis GL. Nonalcoholic fatty liver disease after liver transplantation for cryptogenic cirrhosis or nonalcoholic fatty liver disease. Liver Transpl 2010; 16: 431-439. [ Links ]
130. Nayak NC, Vasdev N, Saigal S, Soin AS. End-stage nonalcoholic fatty liver disease: evaluation of pathomorphologic features and relationship to cryptogenic cirrhosis from study of explant livers in a living donor liver transplant program. Human Pathology 2010; 41: 425-430. [ Links ]
131. Malik SM, Gupte PA, deVera ME, Ahmad J. Liver transplantation in patients with nonalcoholic steatohepatitis-related hepatocellular carcinoma. Clinical gastroenterology and hepatology 2009; 7: 800-806. [ Links ]
132. Argo CK, Caldwell SH. Epidemiology and natural history of nonalcoholic steatohepatitis. Clin Liver Dis 2009; 13: 511-531. [ Links ]
133. Charlton M. Obesity, Hyperlipidemia and Metabolic Syndrome. Liver Transplantation 2009; 15(11): S83-S89. [ Links ]
134. Malik SM, deVera ME, Fontes P, Shaikh O, Ahmad J. Outcome after liver transplantation for NASH cirrhosis. American Journal of Transplantation 2009; 9: 782-793. [ Links ]
135. Pagadala M, Dasarathy S, Eghtesad B, McCullough AJ. Posttransplant metabolic syndrome: an epidemic waiting to happen. Liver Transpl 2009; 15: 1662-1670. [ Links ]
136. Koehler E, Watt K, Charlton M. Fatty Liver and Liver Transplantation. Clin Liver Dis 2009; 13: 621-630. [ Links ]
137. Dumortier J, Giostra E, Belbouab S, Morard I, Guillaud O, Spahr L, et al. Non-alcoholic fatty liver disease in liver transplant recipients: another story of "seed and soil". Am J Gastroenterol 2010; 105: 613-620. [ Links ]
138. Ong J, Younossi ZM. Non-alcoholic fatty liver disease after liver transplantation: a case of nurture and nature. Editorial. Am J Gastroenterol 2010; 105: 621-623. [ Links ]

