










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.25 no.4 Bogotá Oct./Dec. 2010
Tejido linfoide y linfomas gástricos
Rocío del Pilar López P, MD (1), Rafael Enrique Andrade P, MD (2)
(1) Médica Patóloga. Hospital Universitario Fundación Santa Fe de Bogotá. Profesora Universidad de Los Andes. Bogotá, Colombia.
(2) Médico Patólogo. Jefe Departamento de Patología y Laboratorios Fundación Santa Fe de Bogotá. Profesor Universidad de Los Andes y Universidad Nacional de Colombia. Bogotá, Colombia.
Fecha recibido: 13-09-10 Fecha aceptado: 05-10-10
Resumen
En este artículo hemos realizado una amplia revisión de los linfomas primarios gástricos, su clasificación y aspectos clínico-patológicos más importantes, haciendo énfasis especial en los linfomas MALT o asociados a las mucosas y su relación con la infección por Helicobacter pylori.
Palabras clave
Estómago, tejido linfoide, linfoma primario gástrico, linfomas extranodales, linfoma asociado a mucosas (MALT), linfoma de la zona marginal, linfoma B difuso de célula grande, linfoma del manto, linfoma folicular, Helicobacter pylori.
TEJIDO LINFOIDE EN LA MUCOSA GASTRICA
El tejido linfoide a nivel del tracto gastrointestinal se encuentra disperso en toda su extensión; sin embargo, solamente a nivel del tracto aerodigestivo superior y a nivel del íleon terminal se encuentra organizado en estructuras similares a las presentes en otros órganos linfoides secundarios como el bazo y los ganglios linfáticos. A nivel de la orofaringe constituye el anillo de Waldeyer y a nivel del íleon terminal las placas de Peyer. En condiciones normales, a nivel del esófago y el estómago en donde no hay folículos linfoides establecidos, muy probablemente porque no es necesario a este nivel la organización linfoide en estructuras funcionales por el tránsito intestinal rápido y el medio ambiente hostil para los microorganismos. En condiciones patológicas de tipo inflamatorio, infeccioso o tumoral es posible que un estímulo antigénico persistente favorezca la adquisición de un tejido linfoide organizado en una forma tal que semeje funcionalmente el tejido linfoide asociado a las mucosas normalmente presente en otras localizaciones. Este tejido linfoide en condiciones normales puede verse también a nivel del apéndice cecal.
Los elementos celulares que constituyen el tejido linfoide a nivel de estos órganos están presentes en tres diferentes compartimientos, el primero de ellos corresponde a los agregados linfoides verdaderos de la mucosa con formación de centros germinales, zonas del manto y una corona de linfocitos periféricos denominados linfocitos de la zona marginal, el segundo al tejido linfoide que se localiza disperso en la lámina propia y por último los linfocitos intraepiteliales.
La respuesta inmunológica que se desarrolla en las mucosas es muy similar a la que ocurre en el ganglio linfático. El componente celular presente en la lámina propia es muy heterogéneo con presencia de macrófagos, células dendríticas, células plasmáticas, linfocitos T ayudadores y muy escasos eosinófilos semejando en cierta forma las zonas interfoliculares de los ganglios linfáticos. Los linfocitos intraepiteliales son principalmente linfocitos de fenotipo T citotóxicos/supresores y están presentes más notoriamente en el intestino delgado. Los folículos linfoides son el lugar de amplificación de la respuesta inmune humoral, la cual a este nivel está particularmente comprometida con la producción de inmunoglobulina A (1).
A lo largo de todo el tracto gastrointestinal se pueden encontrar diferentes condiciones que alteran la estructura normal del tejido linfoide, por un lado procesos que llevan a un agrandamiento del tejido linfoide normalmente presente como son los fenómenos hiperplásicos asociados procesos inflamatorios, condiciones que aumentan particularmente las poblaciones de linfocitos T como algunos procesos inmunológicos y, en el estómago, particularmente la infección por Helicobacter pylori y finalmente condiciones en las cuales hay neoformación de un sistema linfoide organizado como es el caso de la infección persistente por Helicobacter pylori en el estómago que resulta en la formación de verdaderos folículos linfoides (2).
Por otro lado, a nivel del tracto gastrointestinal es posible encontrar proliferaciones neoplásicas de cualquiera de los componentes linfoides presentes en forma normal o adquirida. Estos procesos constituyen los linfomas, los cuales pueden ser condiciones primarias (linfomas extranodales) o representar la infiltración del tracto gastrointestinal por un linfoma originado en los ganglios linfáticos (linfomas nodales).
LINFOMAS GASTRICOS
Introducción
Sin lugar a duda el linfoma más frecuente a nivel de la mucosa o pared gástrica es el linfoma extranodal del tejido linfoide asociado a mucosas o linfoma MALT, el cual describiremos en mayor detalle a continuación, otros linfomas que pueden presentarse a nivel gástrico se mencionarán posteriormente tales como el linfoma folicular originado en los linfocitos del centro germinal, los linfomas del manto originados en las células que forman los mantos foliculares, los linfomas difusos de célula grande de origen centrogerminal o en células activadas y con menor frecuencia otros linfomas de fenotipo T. Todos ellos en mayor o menor grado constituirán parte del diagnóstico diferencial de los linfomas MALT así como las condiciones hiperplásicas que se describen dentro del contexto de la gastritis folicular (3, 4).
Definición
Desde los años 60, Dawson propuso una serie de criterios diagnósticos necesarios para definir un linfoma primario del tracto gastrointestinal (TGI); dentro de ellos el más importante es la ausencia de linfadenopatías periféricas, un recuento leucocitario con diferencial normal para excluir estados leucémicos con compromiso gástrico, ausencia de compromiso hepático o esplénico por cualquier proceso linfoproliferativo. Estos criterios son válidos, aunque en forma excepcional y usualmente en forma tardía, neoplasias linfoides originadas en el tracto gastrointestinal, especialmente en el estómago pueden extenderse a órganos linfoides distantes e incluso comprometer la medula ósea o presentar células circulantes.
El linfoma primario gástrico por tanto será aquel que se origine en el estómago con compromiso o no de los ganglios linfáticos contiguos. Debe siempre, como se ha mencionado anteriormente, realizar estudios de extensión para excluir el compromiso secundario por linfomas nodales, el cual puede ocurrir hasta en el 25% de estos y conlleva un pronóstico y un esquema terapéutico diferente (5).
Epidemiología
Cerca del 40% de los linfomas no Hodgkin (LNH) se originan en sitios extranodales, siendo el tracto gastrointestinal una de las localizaciones más frecuentes; en el mundo occidental varía entre el 4 y el 18%, mientras que en el Medio Oriente es aún mayor, cercano al 25%.
En el estómago, los linfomas son relativamente infrecuentes, representan menos del 15% de todos los tumores gástricos y solo el 2% del total de los linfomas; sin embargo, es en el estómago donde se originan casi el 75% de los LNH del tracto gastrointestinal (TGI), y su incidencia ha venido en aumento en los últimos años.
De acuerdo con la Organización Mundial de la Salud (WHO) los más frecuentes son los linfomas B difusos de célula grande (LBDCG), algunos de ellos como parte del proceso de progresión de linfomas asociados a las mucosas (MALT), seguidos en frecuencia linfomas B de la zona marginal asociados al tejido linfoide de las mucosas (MALT) (4).
Subtipos
Linfoma de la zona marginal o linfoma asociado al tejido linfoide de las mucosas (MALT)
Fueron Isaacson y Wright en 1982 los primeros en describir una variante del linfoma B de localización extranodal, localizado especialmente en el tracto gastrointestinal, sugiriendo una histogénesis común en el tejido linfoide asociado a las mucosas (6).
La WHO divide los linfomas B de la zona marginal en nodales y extranodales. Ambos se originan en las células B localizadas en la zona marginal y comparten características morfológicas e inmunofenotípicas, pero los extraganglionares dada su inusual patogenia son considerados una entidad diferente (4).
Los que aquí nos ocupan son los linfomas extranodales asociados al tejido linfoide de las mucosas o llamados linfomas MALT gástricos; estos procesos se originan en una mucosa normalmente carente de un tejido linfoide organizado, el cual se ha desarrollado en forma secundaria a procesos inflamatorios crónicos tales como infecciones crónicas persistentes o trastornos de tipo autoinmune. El ejemplo más conocido relacionado con la primera es la gastritis crónica asociada a la infección por el Helicobacter pylori y como ejemplo de la segunda condición la asociación con enfermedades de tipo autoinmune en glándulas salivares como el síndrome de Sjögren o en el tiroides la tiroiditis de Hashimoto.
En 1991, Wotherspoon y colaboradores fueron los primeros en demostrar la estrecha relación que existe entre el Helicobacter pylori y el linfoma MALT; describieron su presencia en prácticamente todos los pacientes con linfoma MALT (7) y dos años después demostraron la regresión tumoral en 5 de 6 pacientes luego de la erradicación de la bacteria (8).
Por lo tanto, es esperable que haya una mayor incidencia de linfoma en países con mayor prevalencia de la infección, llegando a ser cercana al 100% en los linfomas MALT y entre el 50 y 60% en los linfomas que han progresado a linfoma B difuso de célula grande (LBDCG).
Los pacientes con síndrome de Sjögren o sialoadenitis linfoepitelial tienen un riesgo aumentado hasta 44 veces de desarrollar síndromes linfoproliferativos, estos corresponden solamente al 4-7% de los pacientes con linfoma MALT. Cerca del 85% de los pacientes con estas patologías que tienen linfomas corresponden a linfomas MALT. Los pacientes con tiroiditis de Hashimoto tienen un riesgo 3 veces mayor que la población de desarrollar linfoma y 70 veces más de desarrollar un linfoma en localización tiroidea (9).
Los linfomas asociados a mucosas representan aproximadamente entre el 8 y el 10% de los linfomas B; en el estómago son entre el 40 y 50% de todos los linfomas gástricos. Con una incidencia global estimada de 0,7-0,8 por 100.000 habitantes y entre el 3,8 y 6,3 por 100.000 en algunos países del norte de África. Es considerada una enfermedad crónica, localizada e indolente y para su diagnóstico se requiere una estrecha correlación clínica/endoscópica, morfológica, de inmunofenotipo y de estudios de biología molecular.
La mayoría de los casos se presentan en la edad adulta, con una edad media de 61 años y un discreto predominio en el sexo femenino, (la relación mujer:hombre, es de 1,2:1) (10, 11).
Patogénesis
La infección por Helicobacter pylori inducen en el huésped una intensa respuesta inmune innata que involucra varios componentes del sistema inmune, incluyendo la secreción de péptidos antimicrobianos, del tipo de la proteína Nod1, (importante en la señalización de la activación de las caspasas y útil en la respuesta defensiva del huésped), y la expresión de moléculas proinflamatorias en las células huésped teniendo como vía principal la de los receptores Toll-like.
Esta respuesta inmune resulta en un cuadro clínico patológico de gastritis crónica con la posibilidad de formación de folículos linfoides compuestos por una población predominantemente de células B en varios estadios evolutivos, linfocitos T activados tanto por la acción del H. pylori como por las citoquinas del huésped que estimulan la proliferación de linfocitos B tumorales. En una primera fase, la reacción inflamatoria resulta en una hiperplasia linfoide reactiva, si continúa la activación de los linfocitos B es posible que se suceda una proliferación monoclonal de células linfoides con un aspecto fenotípico que semeja los centrocitos del centro germinal, los cuales se localizan en la periferia del folículo linfoide en la denominada "zona marginal"; posteriormente, si la expansión continúa, estas células penetran la lámina propia e invaden las células epiteliales produciendo una lesión destructiva que semeja el hallazgo histológico visto en las glándulas salivares, el cual se ha denominado "lesión linfoepitelial". Es probable que además de esta expansión celular se suceda en forma asociada una reacción autoinmune indirecta que podría explicar el por qué los linfomas de tipo MALT se localizan y permanecen en la pared gástrica e incluso regresan con la erradicación del H. pylori.
La inmunidad del H. pylori es dependiente de los linfocitos T, participan los linfocitos T CD8 positivos de tipo supresor, los cuales se categorizan por su capacidad de secretar citoquinas y sus funciones citotóxicas; por otro lado, los linfocitos ayudadores Th1 secretan entre otros factor de necrosis tumoral e interferón γ y los linfocitos Th2 interleuquinas IL4,5 y 10 que facilitan la producciσn de anticuerpos por las células B. La respuesta del huésped a las citoquinas es importante en el desarrollo de gastritis o tumor. El patrón de inflamación gástrico va a determinar cuál tipo de enfermedad se presentará, siendo prerrequisito para el desarrollo del linfoma la presencia de inflamación gástrica inducida por H. pylori.
Además de estos factores, también es de gran importancia la capacidad de adaptación del H. pylori al sistema inmune del huésped, que resulta en últimas en la perpetuación de la infección, lo cual va a depender del balance entre los linfocitos Th1/Th2 dado por el papel del ADN genómico recombinante y la expresión variable de antígenos de tipo Lewis de grupo sanguíneo que permiten la unión al receptor DC-SIGN (12).
En relación con la autoinmunidad gástrica y el H. pylori, ha sido descrito que la gran mayoría de los pacientes con gastritis autoinmune ha tenido o tiene infección por H. pylori, induciendo la producción de autoanticuerpos que llevan a la destrucción glandular con pérdida de la producción de las células parietales lo que se traduce en atrofia gástrica en los individuos susceptibles (13).
Por otro lado, también se ha sugerido que pueda existir una predisposición genética en los linfomas gástricos; entre los factores que puedan estar involucrados se ha visto una mayor prevalencia en individuos con haplotipos HLADQA1* 0103, HLA-DQB1*0601 y DQA1*0103-DQB1*0601 (14).
Genética molecular
En la gran mayoría de los pacientes que presenta linfomas de tipo MALT se identifica la presencia de rearreglos en el gen de la cadena pesada de las inmunoglobulinas (IgH); se ha visto que dependiendo de la profundidad de la lesión puede encontrase presencia de oligoclonas (lesiones confinadas a la mucosa) y francas poblaciones monoclonales en masas tumorales y lesiones infiltrativas (15).
En el 50% de los linfomas MALT se encuentran anomalías citogenéticas siendo la más frecuente la traslocación t(11;18), (q21 yq21); los genes involucrados en esta traslocación son el gen AP12 en 11q21 y el gen MLT en 18p21; es observada en el 25 a 50% de los casos. También se han descrito alteraciones del gen Bcl10 con sobreexpresión del mismo asociada a la traslocación t(1;14) (p22;q32) donde participa un nuevo gen llamado MLT1. Ocurre en cerca del 5% de los pacientes. Tanto las translocaciones t(1;14) como la t(11;18) resultan en la activación de la vía de señalización asociada al factor de transcripción esencial NK- κappaB. Estos eventos genéticos parecen específicos y prácticamente solo han sido observados en los linfomas MALT. Se presentan con mayor frecuencia en estadios avanzados y en aquellos casos que no responden a la erradicación del Helicobacter pylori (16).
Además de los eventos mencionados, se han encontrado trisomías del cromosoma 3 (entre el 30 y el 60%) usualmente asociadas a la t(11;18) y menos frecuentemente trisomías de los cromosomas 7,12 y 18; estas últimas dependientes del estímulo antigénico, presentes también en LBDCG. Los eventos genéticos mencionados son mutuamente excluyentes. Otras alteraciones de los genes Bcl6, p16 y mutaciones de la proteína p53 pueden verse asociados a progresión de la lesión a LBDCG, eventos que ocurren en menos del 10% de los casos (17).
Desde el punto de vista del comportamiento biológico y los eventos moleculares mencionados, se han dividido los linfomas gástricos en tres categorías, las cuales pueden tener importancia en cuanto al manejo clínico de los pacientes; estos grupos representan la posibilidad de respuesta a la erradicación del H. pylori y la presencia de la fusión API2-MALT1 resultante de la traslocación t(11,18) (q21:q21). El grupo A corresponde a los linfomas MALT cuya histología no demuestra progresión a alto grado, los cuales presentan clínicamente un compromiso gástrico superficial o un estadio temprano. El grupo C, lesiones en las cuales hay infección leve por H pylori pero no enfermedad clínica avanzada, en los cuales tampoco hay evidencia de progresión histológica pero se encuentra expresión por inmunohistoquímica del Bcl10, y finalmente el grupo B, no muy bien caracterizado aún, en el cual se demuestra extensión a los ganglios linfáticos, infiltración profunda de la pared gástrica indicativa de un estadio clínico avanzado y en el cual hay frecuentemente a nivel histológico un componente acompañante de células grandes. Con base en estos hallazgos, es posible hacer una predicción de la respuesta a la erradicación del H. pylori, la cual se asocia con una alta probabilidad de remisión de la enfermedad. Entre los factores más importantes que permiten predecir una regresión de la enfermedad tras la erradicación de la bacteria están la ausencia de la fusión API2-MALT1 documentada por FISH, el estadio clínico temprano con invasión superficial confinada a la mucosa gástrica y la localización antral (18-20).
Manifestaciones clínicas y hallazgos endoscópicos
Síntomas clínicos como anemia, melenas, hematemesis y pérdida de peso, solo son vistos en menos del 42% de los pacientes con MALT, la mayoría de los linfomas son indolentes o se manifiestan con síntomas dispépticos, sensación de quemadura, dolor abdominal o epigástrico, incluso en pacientes con estadios avanzados de la enfermedad (21).
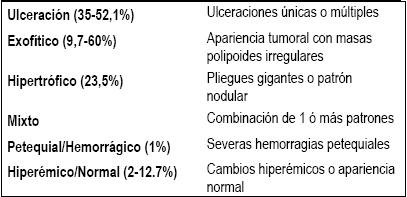
Los hallazgos endoscópicos son el reflejo de la heterogeneidad de esta entidad, presentándose frecuentemente todo un espectro de imágenes. Se describe la clasificación endoscópica en la tabla 1, con porcentajes muy variables en sus presentaciones según los diferentes estudios (22, 23) (tabla 1).
Tabla 1. Presentación endoscópica (16).
Patología
El diagnóstico se basa en el estudio histopatológico, de inmunofenotipo y en la actualidad resulta de gran utilidad la caracterización molecular por técnicas de hibridización in situ con fluoresceína o FISH.
Por tratarse de una entidad con un patrón muy heterogéneo de presentación, como ya fue referido, se recomienda realizar para el diagnóstico inicial múltiples biopsias (entre 8 y 12) de lesiones visibles y al menos 2 biopsias de áreas de aspecto normal tanto a nivel antral como corporal, reservando material fijado en formol tamponado neutro al 10% y de ser posible tejido en fresco para una mejor caracterización fenotípica por citometría de flujo.
En el seguimiento de la enfermedad ya diagnosticada las guías ESMO recomiendan realizar biopsias, igualmente manejadas, a los 2-3 meses postratamiento y posteriormente 2 veces por año, para el monitoreo histológico de la regresión tumoral. En todos los pacientes con enfermedad residual mínima estable o remisión completa se recomienda evaluación endoscópica y monitoreo sistémico anual; más aún teniendo en cuenta que existe un riesgo de desarrollar adenocarcinoma gástrico en pacientes que han tenido un linfoma gástrico previo del tipo MALT (6 veces) ya que ambos procesos neoplásicos comparten en parte mecanismos fisiopatológicos similares (24).
Los hallazgos histopatológicos los podemos dividir en hallazgos morfológicos, hallazgos fenotípicos o inmunofenotípicos y hallazgos moleculares.
Hallazgos morfológicos
Patrón de crecimiento: El infiltrado linfoide característico de esta neoplasia se localiza en las zonas marginales de los folículos linfoides extendiéndose en la lámina propia hacia las regiones interfoliculares. Parte de la dificultad diagnóstica radica en la ausencia frecuentemente de folículos linfoides representados en el material de biopsia, razón por la cual, como se mencionó anteriormente, una amplia representación de la mucosa es deseable.
Hallazgos citológicos: Las células neoplasia en este tipo de linfoma pueden adoptar diferentes características; un grupo importante de lesiones muestra una población celular que semeja las células de la zona marginal normal presentes en ganglios linfáticos, en el bazo y en el tejido MALT normal, caracterizadas por tener un citoplasma amplio claro, bordes bien definidos y un núcleo central en algunas ocasiones indentado o de aspecto monocitoide; otros pacientes presentan, por el contrario, un infiltrado celular monótono a veces indistinguible de las células linfoides pequeñas como es el visto en linfomas de bajo grado de tipo linfocítico o de células del manto nodales y finalmente un infiltrado por células con escaso citoplasma y núcleo hendido semejando los centrocitos normales del centro germinal. Es frecuente la diferenciación plasmocitaria especialmente hacia los tercios superficiales de la mucosa y hasta en una tercera parte de los casos se identifican la presencia de inclusiones nucleares las cuales pueden ser positivas con la coloración de PAS denominadas cuerpos de Dutcher (figura 1). Hay igualmente presencia de algunos inmunoblastos y ocasionales células grandes, usualmente con una muy baja actividad mitótica. Acompañando el infiltrado de células neoplásicas pueden encontrarse frecuentes eosinófilos y neutrófilos, estos últimos a nivel del epitelio glandular.
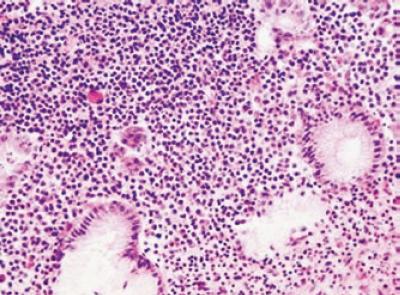
Figura 1. Mucosa con infiltrado inflamatorio monomorfo, con diferenciación plasmocitaria H-E 40x.
El epitelio glandular en los linfomas asociados a mucosas está frecuentemente alterado, por un lado hay expansión linfoide con desaparición de estructuras glandulares bien formadas, dejando la presencia de células residuales atrapadas las cuales sufren un proceso de degeneración eosinofílica citoplasmática. Un hallazgo histopatológico clave e indispensable en el establecimiento inicial del diagnóstico es la presencia de la lesión linfoepitelial definida por la infiltración con destrucción glandular de células linfoides atípicas de aspecto centrocítico en pequeños grupos (figuras 2 y 3) (3, 25).
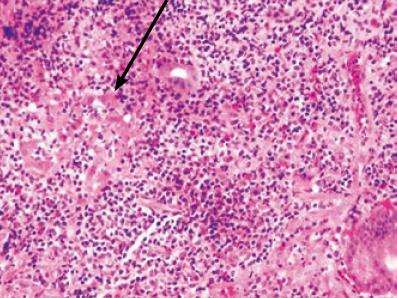
Figura 2. Lesión linfoepitelial. H-E 40X.
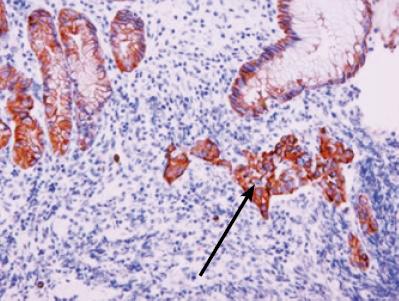
Figura 3. Estudio de inmunohistoquímica para citoqueratina AE1/AE3, identifica la lesión linfoepitelial. 40x.
Los criterios histológicos más importantes y útiles especialmente en el diagnóstico diferencial fueron establecidos por Wotherspoon en 1993 y aún siguen vigentes, se muestran en la tabla 2 y son importantes también en la evaluación del seguimiento y su respuesta terapéutica (4, 26).
Tabla 2. Criterios histológicos establecidos por Wotherspoon (16).
Hallazgos inmunofenotípicos
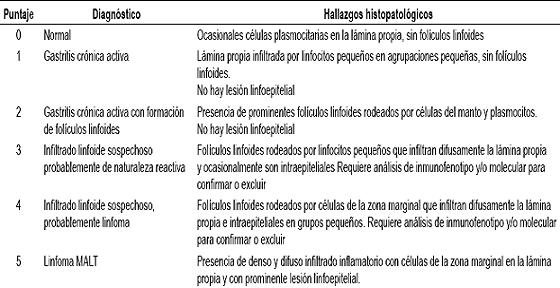
El linfoma MALT se caracteriza por una población heterogénea de células de linaje B. El patrón de inmunofenotipo corresponde al normalmente observado en la zona marginal de un ganglio linfático. Las células tumorales reaccionan para los marcadores pan B CD20 (+), (figura 4), CD79a (+), PAX 5, con negatividad para el CD3; este último es una marcador específico de células T y es el predominante en pacientes con gastritis crónica difusa. No es inusual encontrar en linfomas MALT la coexpresión del CD43, un hallazgo fenotípico aberrante que cuando está presente en una población linfoide de fenotipo B sugiere que se trata de una expansión neoplásica. En grupo menor al 50% de los casos es posible documentar la presencia de restricción clonal con estudios de inmunohistoquímica para las cadenas ligeras κ o λ; sin embargo, dado que este es un linfoma que se origina en un tejido linfoide previamente expandido en forma anormal, no es inusual que no sea posible en las fases tempranas de la enfermedad de documentar la población clonal con esta metodología, de hecho se requiere que por lo menos la relación entre células que expresan las cadenas kappa sobre lambda sea superior a 4:1 ó 5:1 para poder estar seguros de que hay un exceso clonal de células kappa y una delación de 1:1 ó mayor para probar que hay un exceso de células lambda. Las células tumorales usualmente no expresan el CD5, CD23 ni el CD 10, información muy útil para excluir otras neoplasias de células pequeñas y de fenotipo B que entran en el diagnóstico diferencial como son el linfoma de células del manto, el linfoma linfocítico de célula pequeña y el linfoma folicular. Las células tumorales también son reactivas para Bcl2, negativas para el Bcl 6(-) y la Ciclina D1 (-). Anticuerpos como la citoqueratina pueden ser de mucha utilidad especialmente para demostrar la presencia de células epiteliales atrapadas y para confirmar la existencia de la lesión linfoepitelial cuando el infiltrado es muy denso (3, 4).
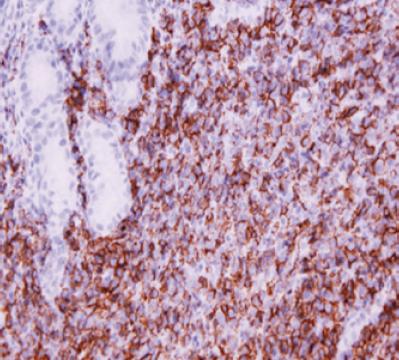
Figura 4. Estudio de inmunohistoquímica para CD20, la reactividad intensa y difusa de las células tumorales demuestra el fenotipo de células B.
En algunos pacientes, los estudios de inmunofenotipo descritos anteriormente no son confirmatorios, son realizados en parafina y en estas circunstancias es deseable la realización de estudios de citometría de flujo en material fresco preservado en solución salina, dado que esta metodología puede ser mucho más sensible en la documentación de aberrancias fenotípicas y ser más útil en la documentación de una expansión clonal anormal.
Hallazgos genéticos y moleculares
Los análisis genéticos en la actualidad están ampliamente recomendados en diferentes circunstancias; por un lado pueden ser de gran utilidad diagnóstica, en algunas ocasiones, especialmente ante la presencia de biopsias pequeñas, con fenotipo incierto o compromiso tumoral focal, los estudios moleculares pueden tener un gran valor si se documenta la presencia de alguna de las anomalías genéticas mencionadas; para estos estudios el mismo material o un nuevo material más representativo puede someterse a pruebas de FISH. Por otro lado, los estudios moleculares son de utilidad, se ha demostrado que la presencia de la traslocación t(11;18) (q21; q21) como factor pronóstico adverso, ya que entre el 20-30% de los pacientes que la poseen, no responden al tratamiento para la erradicación del H. pylori. Considerada la terapia de primera línea requerirán otro tipo de terapia. Además, en los casos t(11;18) negativos, la expresión de FOXP1, determinada por técnicas de inmunohistoquímica y FISH predice igualmente la resistencia a erradicación del H. pylori. La presencia de la proteína BCL10, localizada a nivel nuclear e implicada en la señalización NF- κappaB, observada con estudios de inmunohistoquνmica, puede predecir una respuesta terapéutica. Por lo tanto, realizar estudios adicionales puede constituirse en una alternativa para aquellos casos t(11;18) negativos (27, 28).
En un reciente proyecto realizado en nuestra Institución se demostró con el estudio de 50 casos de linfoma MALT la asociación de gastritis crónica y H. pylori. No se encontró la presencia de t(11;18) (q21,q21), ni transformación a linfoma B difuso de célula grande en el 66% de los casos con regresión tumoral luego de tratamiento antibiótico para la erradicación bacteriana, mientras que el 14% de los pacientes que no tuvo regresión tumoral luego del tratamiento de primera línea sí tenían la mutación, (figura 5) además en todos los que persistió el tumor se encontró expresión de la proteína BCL10 (29).
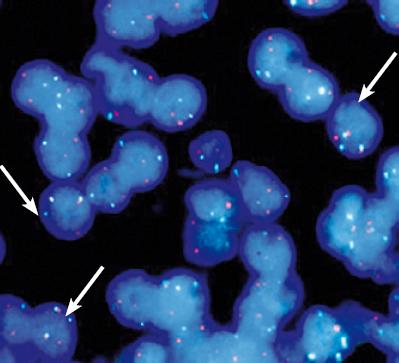
Figura 5. Hibridización in situ fluorescente (FISH) para la detección de la t(11;18) (q21;121) utilizando la sonda LSI API2-MALT1 dual color (Vysis, Downers Grove, IL, USA). Señales de hibridización de la translocación en filtro triple, verde y rojo (flechas) (Foto cortesía Sally Yepes).
Los estudios moleculares de clonalidad han sido controvertidos ya que en pacientes con gastritis foliculares pueden verse expansiones oligoclonales que pueden ser interpretadas en biopsias pequeñas como poblaciones monoclonales. Tienen valor establecido en el seguimiento de la enfermedad al detectarse la misma clona neoplásica que al inicio, especialmente si la enfermedad es endoscópicamente manifiesta, ya que clonas residuales tumorales pueden permanecer durmientes en la mucosa en pacientes que nunca van a presentar una reactivación posterior de su enfermedad.
DIAGNOSTICO DIFERENCIAL
Incluye numerosos procesos reactivos desde la gastritis crónica por Helicobacter pylori, la gastritis folicular, hasta procesos neoplásicos de células B pequeñas como son el linfoma folicular, linfoma de células del manto, linfoma linfocítico de célula pequeña. Su diferenciación se basa no solamente en los hallazgos morfológicos antes descritos sino en un estudio completo de inmunofenotipo.
La demostración de restricción de cadenas livianas es importante en el diagnóstico diferencial con infiltrados linfoides benignos. Al igual que la expresión anormal del CD5 presente tanto en el linfoma linfocítico de célula pequeña como en el linfoma del manto, este último acompañado de la expresión nuclear de la ciclina D1. La presencia de CD10 por otro lado es un marcador de gran ayuda presente en los linfomas de origen centrogerminal o folicular (3, 4).
En pacientes en los cuales no es posible hacer un diagnóstico diferencial conclusivo con procesos de naturaleza reactiva se recomienda la realización de nuevas biopsias en lo posible antes de iniciar la terapia de erradicación del HP.
Pronóstico
La gran mayoría de los linfomas MALT (90%) tiene un curso indolente con muy baja tendencia a la enfermedad diseminada, aun cuando algunos estudios han demostrado un porcentaje mayor cercano al 30% de enfermedad diseminada al momento del diagnóstico, en pacientes o con un estadio avanzado o en aquellos que han presentado múltiples episodios de recurrencia. La sobrevida a 5 años es de aproximadamente del 90% (30).
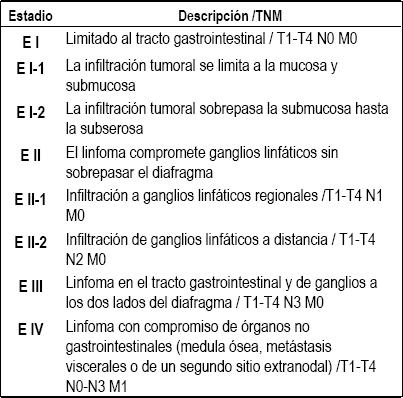
Basado en múltiples estudios prospectivos y multicéntricos se ha establecido que el diagnóstico histopatológico completo y la estadificación basada en la diseñada por Ann Arbor y modificada por Musshof y Radaszkiewicz (tabla 3) que requiere ultrasonido abdominal completo, tomografía computarizada de tórax y abdomen, biopsia de médula ósea, ultrasonido endoscópico completo son considerados los dos factores pronósticos más importantes (16, 31, 32).
Tabla 3. Ann Arbor modificado para el estadiaje (16).
En más de la mitad de los casos hay remisiones prolongadas como respuesta al tratamiento con antibióticos del linfoma inducido por la asociación de gastritis por Helicobacter pylori. Y también en la mitad de los casos que presentan la traslocación t(11;18) (q21;q21) hay resistencia al tratamiento de erradicación del H. pylori, y además su presencia le confiere un efecto protector para la transformación a linfoma difuso de célula grande.
Transformación a linfoma B difuso de célula grande puede ocurrir en menos del 10% de los pacientes, usualmente en relación con un curso prolongado, asociado a otros eventos genéticos como mutaciones del p53(17p13) y deleciones del p16(9p21); no tiene relación con enfermedad diseminada (31).
La terapia de erradicación para el H. pylori es la primera línea terapéutica de elección; en caso de pobre respuesta a esta o ausencia de la bacteria, la radioterapia dirigida al estómago y a los ganglios perigástricos a una dosis de 40Gy está indicada.
LINFOMA B DIFUSO DE CELULA GRANDE (LBDCG)
En el estómago, estos linfomas son indistinguibles de los linfomas que se originan a nivel nodal. El sitio más común de presentación extranodal es el tracto gastrointestinal y en este el estómago. Su histogénesis no está bien determinada. Corresponden a más del 60% de los linfomas primarios gástricos, con una incidencia de 1 por 100.000 en países desarrollados, incidencia que ha venido en aumento. Pueden presentarse en forma primaria o de novo, más frecuente en hombres por encima de los 50 años. Cuando se presentan como transformación de un linfoma MALT (son los llamados secundarios) la edad de presentación es un poco mayor con una media de 68 años, además esta forma se relaciona con la presencia de mutaciones en los genes p53 y Bcl-6, la sobreexpresión de este último se encuentra entre el 60-73% de los LBDCG (22). También se ha demostrado trisomía del cromosoma 2 y alteración del gen c-myc. El rol del H. pylori en el LBDCG es controversial (33).
La presentación clínica incluye síntomas como anorexia, pérdida de peso y dolor abdominal, siendo menos frecuente el sangrado digestivo. Los síntomas B observados en los linfomas nodales son inusuales en esta presentación. Es un linfoma de crecimiento rápido, potencialmente agresivo, aunque habitualmente se presenta como una enfermedad limitada. Macroscópicamente puede verse como una lesión ulcerada grande muy similar a un adenocarcinoma gástrico avanzado, como úlceras poco profundas o también como una lesión levantada; en contraste con los linfomas MALT, el LBDCG se presenta más como lesión única (78%). Puede o no estar asociada a infección por H. pylori. En las formas asociadas existen algunos casos reportados en la literatura con remisión posterior a la erradicación del microorganismo. El estadio clínico y la profundidad de la invasión son los más importantes factores predictivos de remisión completa o de presencia de enfermedad metastásica a ganglios regionales; si hay compromiso únicamente de mucosa y/o submucosa el compromiso ganglionar es del 3%, mientras si se extiende hasta la muscular propia o más profundamente es del 45 y 73% respectivamente (34, 35).
Morfológicamente, este linfoma se caracteriza por un crecimiento expansivo y destructivo de la arquitectura glandular, la mucosa normal es progresivamente reemplazada por células linfoides grandes, transformadas (figura 6). Citológicamente puede corresponder a lesiones de aspecto heterogéneo; la variante centroblástica es la forma más común y se caracteriza por la presencia de células grandes ovales con nucleolos periféricos que semeja las células grandes no hendidas normalmente presentes en el centro germinal; presentan frecuentes figuras mitóticas indicando agresividad biológica. Estos linfomas pueden o no acompañarse simultáneamente de zonas en las cuales se identifica la presencia de un linfoma MALT sugiriendo que se trata de la progresión del mismo, en esta circunstancia debe diagnosticarse como dos procesos diferentes acorde con las recomendaciones de la OMS (4). El estudio de inmunofenotipo demuestra la expresión de marcadores de células B como CD20, CD19, CD79a y CD22. Puede observarse coexpresión CD5 y CD10, y en la mitad de los casos hay reactividad para Bcl-2 (4).
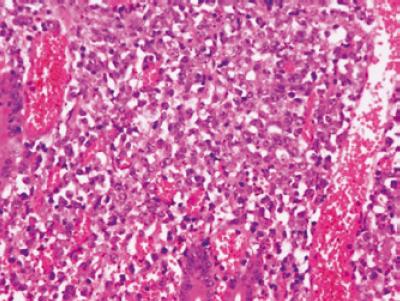
Figura 6. Las células tumorales grandes tienen un crecimiento expansivo y destructivo de la arquitectura glandular. H-E 40x
La expresión de Bcl-6 es encontrada en una alta proporción de casos y la demostración de su traslocación se ha reportado asociada a un mejor pronóstico, mientras que aquellos con presencia de la mutación del p53 tienen una connotación de pobre pronóstico, al igual que altas ratas de proliferación celular (figura 7).
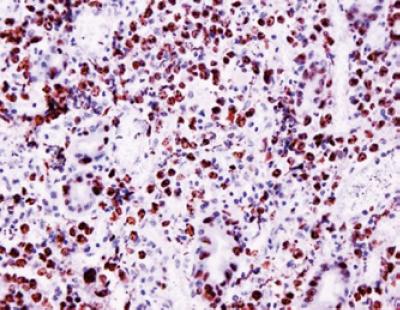
Figura 7. Estudio de inmunohistoquímica para el marcador de proliferación celular Ki67 mostrando un alto índice de proliferación.
El tratamiento convencional con terapia sistémica, sola o en combinación con radioterapia es de elección, en algunos estudios asociada a inmunoterapia, con una respuesta del 95% a 5 años (36).
LINFOMA BURKITT (LB)
El linfoma de Burkitt es un linfoma B no Hodgkin (NHL) altamente agresivo que suele presentarse frecuentemente en sitios extraganglionares o en menor frecuencia como una leucemia aguda; desde el punto de vista epidemiológico puede mostrarse en forma esporádica o asociado a inmunodeficiencias o inmunosupresión, siendo endémico en zonas geográficas como África ecuatorial (37).
Es rara su presentación como linfoma primario gástrico, su incidencia aún no ha sido estimada claramente, pero sigue siendo muy baja comparada con otros linfomas gástricos, es indistinguible morfológica e inmunofenotípicamente de los casos observados en otras localizaciones. Se presenta con mayor frecuencia en hombres, con una edad media de 48 años (rango entre 7 y 75 años), principalmente es visto en una localización corporoantral, en estadios tempranos cuando la enfermedad es ubicada al momento de su presentación; estas lesiones pueden tener una respuesta favorable a la quimioterapia sistémica en al menos el 70% de los casos. Ha sido descrita la posible asociación con H. pylori, especialmente en la edad pediátrica e incluso hay reportes de remisión posterior al tratamiento de erradicación (38).
Al igual que los LDCG, esta neoplasia tiene un crecimiento infiltrativo y destructivo. La característica morfológica más importante es la presencia de una alta proliferación celular acompañada de muerte celular por apoptosis, dando el aspecto conocido como patrón en "cielo estrellado" el cual no es exclusivo de este proceso neoplásico; la proliferación está dada por células linfoides de tamaño mediano, con núcleos redondeados y escaso citoplasma, acompañadas por macrófagos. Los estudios de inmunohistoquímica demuestran fenotipo de células B (CD20, CD19, CD79a o CD22), hay expresión de CD10 y Bcl6 que manifiestan un origen centro germinal de las células tumorales, no expresan Bcl2, CD5, CD23 ni TdT. Tienen característicamente un índice de proliferación celular determinado con el Ki67 muy alto, cercano al 100% (3, 4).
Todos los casos muestran la traslocación del gen MYC (8q24/MYC) de la cadena pesada Ig en el cromosoma 14 t(8;14), menos comúnmente compromete las cadenas livianas lambda 22q11 t(8;22) o kappa 2p12 t(2;8).
Los virus del Epstein Barr (EBV) y la malaria han sido reconocidos como cofactores importantes, en especial en el LB endémico. El genoma EBV se ha demostrado en casi todos los casos endémicos y entre el 25-40% de los casos asociados a inmunodeficiencia, pero el exacto papel del EBV en la patogénesis del LB no está bien entendido aún. Secuencias de este virus son en la actualidad fácilmente detectables por técnicas de hibridización in situ calorimétricas denominadas "ISH" aplicables a tejidos rutinarios fijados en formol y embebidos en parafina.
La estadificación de esta neoplasia se determina según la propuesta establecida por Murphy y Husu, modificada por Magrath que identifica los pacientes con enfermedad limitada o con extensión intrabdominal o intratorácica. El manejo con quimioterapia intensiva y de corta duración conlleva a un buen pronóstico con sobrevidas libres de enfermedad en el 80-90% de los pacientes. En estadios avanzados, e incluso con infiltración a medula ósea o SNC disminuye a un 60% (39).
LINFOMA DE CELULAS DEL MANTO (LCM)
El linfoma de células del manto corresponde entre el 3-10% de los linfomas no Hodgkin, de los cuales entre el 20-30% se originan en el tracto gastrointestinal, a nivel gástrico, es característicamente un componente de la poliposis linfomatoide múltiple del tracto gastrointestinal, siendo muy infrecuente por fuera de este contexto clínico. Clínicamente se manifiesta con dolor abdominal, diarrea, hematoquezia, melenas y fatiga. Puede verse endoscópicamente como una lesión que semeja una gastritis leve en cerca del 40% de los casos o como micropólipos en los restantes. La localización puede ser única o multicéntrica. Afecta con mayor frecuencia a los hombres después de la quinta o sexta década de la vida (40).
El patrón de crecimiento es infiltrativo, aunque puede progresivamente ir reemplazando la mucosa no es inusual ver extensión entre estructuras glandulares especialmente en la periferia de la lesión. Las formas polipotes presentan un patrón de crecimiento claramente nodular.
Morfológicamente se caracteriza por una población de células linfoides monomórficas pequeñas, que semejan los centrocitos o células hendidas de los centros germinales (figura 8), el índice mitótico puede ser alto y cuando está presente es considerado como un factor pronóstico adverso. Fenotípicamente las células tumorales en este linfoma son B, con expresión del CD20 (+) y típicamente coexpresión del marcador T CD5, usualmente CD10 y Bcl6 son negativos. Todos los casos expresan Bcl2 y como criterio indispensable para el diagnóstico está la detección por métodos de inmunohistoquímica de la sobreexpresión de la proteína ciclina D1 (figura 9) como resultado de la traslocación t(11;14) (q13;q32), demostrada en la gran mayoría de los casos; este es un marcador altamente específico de linfoma de células del manto y juega un papel muy importante en su patogénesis. Están descritas algunas variantes inusuales como la forma blastoide de mayor agresividad biológica, en la cual se encuentra mutaciones adicionales en el TP53, p16 y p18. Casi todos muestran sobreexpresión del mRNA para la ciclina D1. La inmunohistoquímica para ciclina D1 parece ser más sensible para el diagnóstico del LCM que los estudios de FISH en la detección de la traslocación t(11;14); sin embargo, frecuentemente en los casos negativos es indispensable realizar estos estudios para poder confirmar el diagnóstico de la enfermedad. El pronóstico de esta neoplasia en la actualidad es pobre, con sobrevida global menor a 3-5 años. La terapia sistémica agresiva sola o combinada con inmunoterapia es el tratamiento de elección (2, 40, 41).
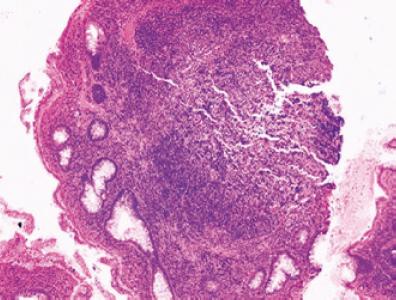
Figura 8. Población de células linfoides monomórficas pequeñas, con crecimiento expansivo que reemplaza la mucosa y separa estructuras glandulares. H-E 10x
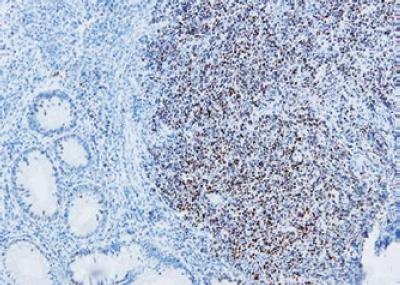
Figura 9. Estudio de inmunohistoquímica muestra reactividad de los núcleos de las células tumorales para la ciclina D1. 20x
LINFOMA FOLICULAR (LF)
Son linfomas que se presentan muy rara vez en forma extranodal; en el tracto gastrointestinal constituyen menos del 3% de los linfomas primarios. Hacen parte frecuentemente del diagnóstico diferencial de procesos reactivos como la hiperplasia folicular del tejido linfoide asociado a las mucosas y en menor frecuencia los linfomas MALT.
Este proceso neoplásico muestra frecuentemente un patrón nodular o folicular de crecimiento; la población celular tumoral esta constituida por células linfoides que semejan la contraparte normal presente en el centro folicular; de esta forma, puede verse la presencia de células pequeñas o centrocitos entremezclados con células grandes o centroblastos; en general, su graduación y variantes están dadas por la cantidad de células de tipo centroblástico y del porcentaje estimado de crecimiento guardando el patrón folicular. La mayoría de células del LF expresan la proteína BCL2, la cual puede ser de ayuda en la diferenciación con las hiperplasias reactivas. Las células tumorales expresan marcadores de células B (CD20, CD79a, CD19, CD22), además son positivas para Bcl6 y CD10 (4).
La anomalía citogenética más frecuente corresponde precisamente a la que compromete el gen del BCL2, en este caso resultante en la traslocación t(14;18)(q32;q21) presente en el 70-95% de los casos nodales y con menor frecuencia en las formas extranodales.
El grado histológico se correlaciona con el pronóstico, siendo los grados 1 y 2 indolentes, razón por la cual se han agrupado estos dos en una categoría general de bajo grado, según los criterios actuales de la OMS (4) y los grado 3 agresivos o de alto grado. No obstante la graduación histológica, son linfomas que frecuentemente responden a la quimioterapia lográndose una remisión completa de la enfermedad y son potencialmente curables con quimioterapia sistémicas; sin embargo, no es inusual la recurrencia de la enfermedad y hasta en un 35% de los casos transformación o progresión a linfomas de mayor agresividad biológica como el LBDCG, este último con la posibilidad de tener de curso clínico agresivo con rápida progresión y muerte, cuando es refractario al tratamiento (42).
LINFOMA DE CELULAS T
Son neoplasias muy infrecuentes a nivel sistémico y a nivel extranodal en países occidentales, representan menos del 7% de los linfomas gástricos. Su etiología es desconocida y la mayoría de ellos reportados en relación con infecciones, asociados con HTLV-1, con el H. pylori y el virus de Epstein Barr (EBV); algunos de los casos vistos en zonas endémicas para el HTLV1 probablemente representen la manifestación gástrica del linfoma/leucemia de células T del adulto (43).
Desde el punto de vista histológico, los linfomas T en las mucosas representan dos formas, una probablemente originada en las células T del tejido linfoide asociado a las mucosas donde las células T de fenotipo ayudador migran al estómago por un estímulo no conocido, estas células T tienen el fenotipo CD3+,CD4+/CD8-/CD103-. La otra forma se origina en los linfocitos T intraepiteliales similar a lo observado en los linfomas T intestinales asociados a enteropatía o enfermedad celiaca; estas neoplasias presentan un fenotipo de células T citotóxicas supresoras CD3+, CD4-/ CD8+ o CD4-/CD8-, con expresión de CD103 y/o CD56. En la mayoría de los reportes los linfomas T corresponde citológicamente a linfomas de células grandes que obligan a descartar en el diagnóstico diferencial un LBDCG; cuando se trata de variantes de célula pequeña el diagnóstico diferencial es con el linfoma MALT (44).
El análisis genético demuestra reordenamiento monoclonal en los genes del receptor gamma (TCRgamma) y beta de las células T. La combinación de terapia sistémica y tratamiento local con resección radical y/o radioterapia es la mejor estrategia para el tratamiento del linfoma gástrico de células T localizado (45).
LINFOMA DE HODGKIN
Es extremadamente raro, representando menos del 1% de los linfomas gástricos, con muy pocos casos descritos en la literatura. Su diagnóstico conlleva gran dificultad diagnóstica; se basa en el reconocimiento histopatológico de las células de Reed Sternberg en la microscopía de luz, las cuales en una biopsia pueden ser muy escasas. Por otro lado y otros linfomas especialmente de células T con expresión de la molécula CD30 pueden presentar células que semejan esta morfología. El diagnóstico en muestras endoscópicas es muy difícil ya que puede quedar representado solamente el componente inflamatorio y semeja una gastritis no específica o una úlcera péptica; la presentación como masa es inusual.
La asociación epidemiológica y patogénica con el virus del Epstein-Barr está bien establecida, la expresión de la proteína LMP1 puede ser encontrada en los tejidos.
El pronóstico es pobre, entre 45-60% de los pacientes fallecen en el primer año luego del diagnóstico (46).
REFERENCIAS
1. Bienenstock J, Ernst PB, Underdown BJ. The gastrointestinal tract as an immunologic organ--state of the art. Ann Allergy 1987; 59: 17-20. [ Links ]
2. Lundgren A, Trollmo C, Edebo A, Svennerholm AM, Lundin BS. Helicobacter pylori-specific CD4+ T cells home to and accumulate in the human Helicobacter pylori-infected gastric mucosa. Infect Immun 2005; 73(9): 5612-9. [ Links ]
3. Hamilton SR, Aaltonen LA. World health organization classification of tumours Pathology and genetics. Tumours of the digestive system. IARC Press Lyon 2000. [ Links ]
4. Swerdlow SH, Campo E, Harris NL, et al. World health organization classification of tumours of haematopoietic and lymphoid tissues. IARC Press Lyon 2008. [ Links ]
5. Tedeschi L, Romanelli A, Dallavalle G, Tavani E, Arnoldi E, Vinci M, Mortara G, Bedoni P, Labianca R, Luporini G. >Stages I and II non-Hodgkins lymphoma of the gastrointestinal tract. Retrospective analysis of 79 patients and review of the literature. J Clin Gastroenterol 1994; 18(2): 99-104. [ Links ]
6. Isaacson PG, Wright DH. Malignant lymphoma of mucosa- associated lymphoid tissue: a distinctive type of B-cell lymphoma. Cancer 1982; 52: 1410-6. [ Links ]
7. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG. Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 1991; 338: 1175-1176. [ Links ]
8. Wotherspoon A, Doglioni C, Diss T, et al. >Regression of primary low-grade B-cell gastric lymphoma of mucosa-associated lymphoid tissue type after eradication of Helicobacter pylori. Lancet 1993; 342: 575-577. [ Links ]
9. Inagaki H. Mucosa-associated lymphoid tissue lymphoma: molecular pathogenesis and clinicopathological significance. Review. Pathol Int 2007; 57(8): 474-84. [ Links ]
10. Ferry, A. Extranodal Lymphoma Arch Pathol Lab Med 2008; 132(4): 565-578. [ Links ]
11. Psyrri A, S. Papageorgiou, T. Economopoulos. >Primary extranodal lymphomas of stomach: clinical presentation, diagnostic pitfalls and management. Annals of Oncology 2008; 19: 1992-1999. [ Links ]
12. Bergman M, DElios M. Review Article. Cytotoxic T Cells in H. pylori-related gastric autoimmunity and gastric lymphoma. Journal of Biomedicine and Biotechnology 2010; Article ID 104918, 10 pages. [ Links ]
13. MP Bergman, CMJE vandenbroucke-Grauls, B. J. Appelmelk, et al. >The story so far: Helicobacter pylori and gastric autoimmunity. International Reviews of Immunology 2005; 24: 63-91. [ Links ]
14. Kawahara Y, Mizuno M, Yoshino T, Yokota K, Oguma K,Okada H, Fujiki S, Shiratori Y. HLA-DQA1*0103-DQB1*0601 haplotype and Helicobacter pylori-positive gastric mucosa associated lymphoid tissue lymphoma. Clin GastroenterolHepatol 2005; 3: 865-868. [ Links ]
15. Lo WY, Li JY, Lai LS, Yeung YW, Lo ST, Tsui WM, Ng CS, Chan. Instability of clonality in gastric lymphoid infiltrates: a study with emphasis on serial biopsies. Am J Surg Pathol 2005; 29: 1582-92. [ Links ]
16. Zullo A, Hassan C, Cristofari F, Perri F, Morini S. Gastric low-grade mucosal-associated lymphoid tissue lymphoma: Helicobacter pylori and beyond. World J Gastrointest Oncol 2010; 2(4): 181-186. [ Links ]
17. Wündisch T, Kim TD, Thiede C, Morgner A, Alpen B, Stotle M, Neubauer A. Etiology and therapy of Helicobacter pylori-associated gastric lymphomas. Annals of Hematology 2003; 82: 535-545. [ Links ]
18. Inagaki H, Nakamura T, Li C, et al. >Gastric MALT lymphoma are divided into three groups based on responsiveness to Helicobacter pylori eradication and detection of AP12 MALT fusion. Am J Surg Pathol 2004; 28: 1560-1567. [ Links ]
19. Wang g, Auerbach A, Wei M, Dow N, Barry TS, Hodge L, Schaffer D, Sobin LH, Aguilera N. t(11;18)(q21;q21) in extranodal marginal zone B-cell lymphoma of mucosa-associated lymphoid tissue in stomach: a study of 48 cases. Modern Pathology 2009; 22: 79-86. [ Links ]
20. Eva M. Murga Penas, et al. >The translocations t(6;18;11)(q24;q21;q21) and t(11;14;18)(q21;q32;q21) lead to a fusion of the API2 and MALT1 genes and occur in MALT lymphomas. Haematologica 2007; 92: 405-407. [ Links ]
21. Zullo A, Hassan C, Andriani A, Cristofari F, Cardinale V, Spinelli GP, Tomao S, Morini S. Primary Low-grade and High grade Gastric MALT-lymphoma Presentation: A Systematic Review. J Clin Gastroenterol 2009. [ Links ]
22. Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98: 975-98. [ Links ]
23. Fischbach W, Dragosics B, Koelve-Goebeler ME, et al. Primary gastric B-cell lymphoma: results of a prospective multicenter study. Gastroenterology 2000; 119: 1191-202. [ Links ]
24. Zucca E, Dreyling M. On behalf of the ESMO Guidelines Working Group. Gastric marginal zone lymphoma of MALT type: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Annals of Oncology 2010; 21 (Supplement 5): v175-v176. [ Links ]
25. Julia´n Arista-Nasr, et al. >Histologic Changes of the Gastric Mucosa Associated with Primary Gastric Lymphoma in Endoscopic Biopsy Specimens. Arch Pathol Lab Med 2000; 124: 1628-1631. [ Links ]
26. Hummel M, Oeschger S, Barth TF, Loddenkemper C, Cogliatti S B, Marx A, Wacker H H, Feller A C, Bernd HW, Hansmann ML, Stein H, Moller P. Wotherspoon criteria combined with B cell clonality analysis by advanced polymerase chain reaction technology discriminates covert gastric marginal zone lymphoma from chronic gastritis. Gut 2006; 55: 782-787. [ Links ]
27. Tousseyn T, Haesevoets E, Geboes K, >De Hertogh G, Van Cutsem E, Sagaert X. (Analysis of prognostic value of biomarkers in gastric MALT lymphomas. Journal of Clinical Oncology, 2010 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2010; 28(15) (May 20 Supplement). [ Links ]
28. Sagaert X, Van Cutsem E, De Hertogh G, Van Loo P, De Wolf-Peeters C, Geboes K, Tousseyn T. Microarray analysis of t(11;18)(Q21;Q21)-positive and -negative gastrointestinal MALT lymphomas. Journal of Clinical Oncology, 2010 ASCO Annual Meeting Proceedings (Post-Meeting Edition) 2010; 28(15) (May 20 Supplement). [ Links ]
29. Yepes S, Torres MM, Saavedra C, Andrade RE. Determinación de la translocación (11;18)(q21;q21) y la expresión del gen BCL10 y su relación con Helicobacter pylori en linfomas MALT gástricos. Presentado como tesis maestría en genética Humana. Universidad Nacional de Colombia. [ Links ]
30. Thieblemont, et al. Mucosa-associated lymphoid tissue lymphoma is a disseminated >disease in one third of 158 patients analyzed. Blood 2000; 95(3). [ Links ]
31. Fischbach W, Goebeler-Kolve M-E, Dragosics B, Greiner A, Stolte M. Long term outcome of patients with gastric marginal zone B cell lymphoma of mucosa associated lymphoid tissue (MALT) following exclusive Helicobacter pylori eradication therapy: experience from a large prospective. Gut 2004; 53: 34-37. [ Links ]
32. Ahmad A, Govil Y, Frank BB. Gastric mucosa-associated lymphoid tissue lymphoma. Am J Gastroenterol 2003; 98: 975-986. [ Links ]
33. Jai Hyang Go, Woo Ick Yang, Howe J Ree, M. Mutational Analysis of the 5 Noncoding Region of the Bcl-6 Gene in Primary Gastric Lymphomas. Mod Pathol 2001; 14: 410-414. [ Links ]
34. Luigi Cavanna L, Pagani R, SeghiniP, Zangrandi A and Paties C. High grade B-cell gastric lymphoma with complete pathologic remission after eradication of helicobacter pylori infection: Report of a case and review of the literature. World Journal of Surgical Oncology 2008; 6: 35. [ Links ]
35. Yoshino T, Omonishi K, Kobayashi K, Mannami T, Okada H, Mizuno M, Yamadori I, Kondo E, Akagi T. Clinicopathological features of gastric mucosa associated lymphoid tissue (MALT) lymphomas: high grade transformation and comparison with diffuse large B cell lymphomas without MALT lymphoma features. J Clin Pathol 2000; 53: 187-190. [ Links ]
36. Leopardo D, Di Lorenzo G, De Renzo A, Federico P, Luponio S, Buonerba C, et al. >Efficacy of rituximab in gastric diffuse large B cell lymphoma patients. World J Gastroenterol 2010; 16: 2526-2530. [ Links ]
37. Chieng J, Garrett J, Ding EL, Sullivan M. Clinical presentation and endoscopic features of primary gastric Burkitt lymphoma in childhood, presenting as a protein-losing enteropathy: a case report. Journal of Medical Case Reports 2009; 3: 7256. [ Links ]
38. Yeon Hee Pak, Won Seog Kim, Hye Jin Kang, Im Il Na, Baek-Yeol Ryoo, Sung Hyun Yang, et al. >Gastric Burkitt lymphoma is a distinct subtype that has superior outcomes to other types of Burkitt lymphoma/leukemia. Annals of hematology 2006; 85(4): 285-90. [ Links ]
39. Baumgaertner I, Copie-Bergman C, Levy M, Haioun C, Charachon A, Baia M, Sobhani I, Delchier J. Complete remission of gastric Burkitts lymphoma after eradication of Helicobacter pylori. World J Gastroenterol 2009; 15(45): 5746-5750. [ Links ]
40 .Brizova H, Kalinova M, Krskova L, Mrhalova M, Kodet R. >Quantitative measurement of cyclin D1 mRNA, a potent diagnostic tool to separate mantle cell lymphoma from other B-cell lymphoproliferative disorders. Diagn Mol Pathol 2008; 17(1): 39-50. [ Links ]
41. Salar, et al. Gastrointestinal involvement in mantle cell lymphoma: a prospective clinic, endoscopic, and pathologic study. Am J Surg Pathol 2006; 30(10): 1274-80. [ Links ]
42. LeBrun D, Kamel O , Cleary ML, Dorfman RF, Warnk R. Follicular Lymphomas of the Gastrointestinal Tract Pathologic Features in 31 Cases and bcl-2 Oncogenic Protein Expression. American Journal of Pathology 1992; 140(6): 1327-1335. [ Links ]
43. Holanda D, Zhao M, Rapoport A, Garofalo M, Chen Q, Zhao X. Primary gastric T cell lymphoma mimicking marginal zone B cell lymphoma of mucosa-associated lymphoid tissue. J Hematopathol 2008; 1: 29-35. [ Links ]
44. Sugita S, Iijima T, Furuya S, Kano J, Yanaka A, Ohta K, Kojima H, Noguchi. Gastric T-cell lymphoma with cytotoxic phenotype. Pathol Int 2007; 57(2): 108-14. [ Links ]
45. Kawamoto K, Nakamura S, Iwashita A, Watanabe J, Oshiro Y, Nakayama Y, Nimura S, Kimura N, Aoyagi K, Yao T, Kuramochi S, Matsuyama A, Kurihara K, Ohshima K, Takeshita M. Clinicopathological characteristics of primary gastric T-cell lymphoma. >Histopathology 2009; 55(6): 641-53. [ Links ]
46. Hossain, Koak Y, Khan FH. Primary gastric Hodgkins lymphoma. World Journal of Surgical Oncology 2007; 5: 119. [ Links ]

