










Services on Demand
Journal
Article
 text in
text in  Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.27 no.2 Bogotá Apr./June 2012
Alteraciones de la coagulación en cirrosis, viejos y nuevos paradigmas
Old and new paradigms for cirrhosis associated coagulation abnormalities
Yesid Alberto Saavedra González, Est.(1), Laura Margarita Ovadía Cardona (1), Octavio Germán Muñoz Maya, MD (2), Gonzalo Correa Arango, MD. (2)
(1) Estudiante de décimo semestre de Medicina, Facultad de Medicina, Universidad de Antioquia, Grupo de Gastrohepatología, Universidad de Antioquia, Medellín, Colombia
(2) Médico internista especialista en Hepatología, Hospital Pablo Tobón Uribe, Grupo de Gastrohepatología, Universidad de Antioquia, Medellín, Colombia
Fecha recibido: 06-12-11 Fecha aceptado: 15-05-12
Resumen
Introducción: el hígado cumple múltiples funciones en la coagulación sanguínea. En enfermedad hepática, por ejemplo, se altera la hemostasis sanguínea, situación que se manifiesta como hipercoagulabilidad en algunos pacientes o sangrado excesivo en otros.
Propósito: revisar la evidencia actual sobre alteraciones en la coagulación de los pacientes con cirrosis.
Métodos: se llevó a cabo una búsqueda utilizando los términos "MESH": blood coagulation disorders, cirrhosis, hemostasis, hypercoagulability, bleeding y blood coagulation tests. Se escogieron aquellos artículos cuyos títulos y abstracts se adaptaran mejor al propósito de esta revisión.
Resultados: de 146 artículos se seleccionaron 76 para la elaboración del presente escrito.
Conclusión: a pesar de las diversas alteraciones que presentan los pacientes cirróticos en la coagulación, el sistema hemostásico se encuentra en un nuevo balance que las pruebas de coagulación utilizadas rutinariamente no reflejan; es por esto que no son herramientas útiles para la predicción del riesgo de sangrado o de complicaciones trombóticas.
Palabras clave
Trastornos de la coagulación, cirrosis, hipercoagulabilidad, sangrado, pruebas de coagulación sanguínea.
INTRODUCCION
El hígado cumple múltiples roles en la coagulación sanguínea, tanto en la hemostasis primaria como secundaria (1). Es el lugar donde se sintetiza el fibrinógeno y todos los factores de la coagulación II, V, VII, IX, X, XI, XII y XIII, excepto el factor VIII, que es sintetizado principalmente por las células endoteliales sinusoidales y, en menor cantidad, por células endoteliales del pulmón, riñón, bazo y cerebro (2, 3).
El sistema de la coagulación consta de un complejo balance entre componentes procoagulantes, anticoagulantes y sistema fibrinolítico que funcionan en el proceso hemostásico y ofrecen una rápida respuesta a la lesión endotelial con depósito de fibrina, agregación plaquetaria y formación del coágulo. En el individuo sano, los eventos de la coagulación están contrarrestados por la actividad anticoagulante que previene la extensión inapropiada del coágulo, minimiza la isquemia local y promueve la lisis de este una vez se ha alcanzado la hemostasis. En el contexto de la enfermedad hepática, la alteración de este balance lleva a la dominancia de alguno de los dos componentes, lo que puede manifestarse como hipercoagulabilidad en algunos pacientes o sangrado excesivo en otros (4).
MATERIALES Y METODOS
Se realizó una búsqueda de artículos de revisión y artículos originales sobre cirrosis y trastornos de la coagulación en las bases de datos Pubmed y Scielo. Se utilizaron los términos "MESH": blood coagulation disorders, cirrhosis, hemostasis, hypercoagulability, bleeding y blood coagulation tests. De los resultados se escogieron aquellos artículos cuyos títulos y abstracts se adaptaran mejor al propósito de esta revisión. Se analizó un total de 146 resúmenes, de los cuales se seleccionaron 72 para su lectura completa. La información obtenida de estos se complementó con artículos referenciados, a partir de los cuales se construyó el presente escrito.
RESULTADOS
Nuevo modelo de la coagulación
Clásicamente el modelo de la coagulación se ha planteado como una "cascada" enzimática que consta de una serie de etapas secuenciales, en las que la activación de un factor de la coagulación estimula al siguiente y cuya finalidad última es la producción de fibrina, componente estructural del coágulo (5). Este es el conocido modelo de las vías intrínseca y extrínseca de la coagulación. A pesar de la aceptación que dicho modelo ha tenido durante mucho tiempo, el papel fundamental del componente celular ha sido descrito en estudios recientes y ahora es bien entendido que la hemostasis no es posible sin la participación de las plaquetas. Adicionalmente se ha demostrado que diferentes tipos de células expresan proteínas procoagulantes, anticoagulantes y receptores para múltiples componentes de la hemostasis, lo que ha supuesto un nuevo paradigma para explicar las reacciones que tienen lugar durante el proceso hemostásico (6).
Según la visión actual, la coagulación se produce en tres etapas interrelacionadas (figura 1):
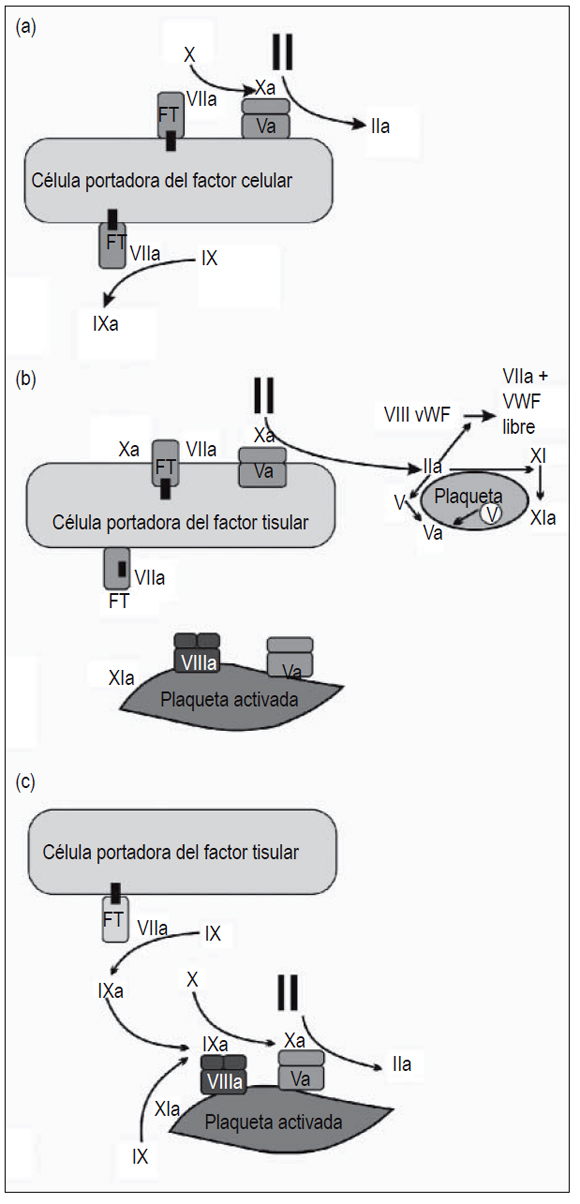
Figura 1. Modelo celular de la coagulación. Modelo celular de la coagulación: (a) Iniciación; (b) amplificación; y (c) propagación (tomada y modificada de: Hoffman M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev 2003; 17(Suppl 1): S1-5).
Fase 1 de iniciación: el factor tisular (FT), un componente integral de la membrana celular, es el principal iniciador de la coagulación in vivo. Se expresa en numerosos tipos celulares como los monocitos circulantes, neutrófilos, fibroblastos y en células endoteliales (7). Cuando se presenta una lesión vascular y la sangre entra en contacto con el subendotelio, se da la interacción del FT celular con el factor VII circulante y lo activa. Este complejo (FT-VIIa) activa posteriormente los factores IX y X. El factor Xa se combina con el factor Va, proceso que ocurre en la superficie celular y da como resultado la producción de trombina en pequeñas cantidades, las cuales participarán en la activación de las plaquetas y del factor VIII en la siguiente fase (8).
Fase de 2 de amplificación: la lesión vascular permite que las plaquetas y componentes plasmáticos entren en contacto con tejidos extravasculares. Una vez esto sucede, las plaquetas se adhieren a la matriz subendotelial y se activan en aquellos sitios donde el FT ha sido expuesto. La trombina generada en la fase 1 amplifica la señal procoagulante al actuar como activador de los factores V, VIII y XI, los cuales se ubican en la superficie plaquetaria para servir como mediadores en reacciones de la siguiente fase (9, 10).
Fase 3 de propagación: esta fase está caracterizada por dos fenómenos principales. En el primero, el complejo tenaza (factor VIIIa, factor IXa, calcio [Ca++] y fosfolípidos) cumple la función catalizadora al promover la conversión del factor Xa; en el segundo, el complejo protrombinasa (factor Xa, factor Va, Ca++ y fosfolípidos) promueve la generación de grandes cantidades de trombina, las cuales son necesarias para la formación de un coágulo resistente a la fibrinólisis. Esto último es lo que se ha denominado explosión de trombina. El complejo protrombinasa es fundamental para la etapa final de esta fase gracias a que es 300 000 veces más activo que el factor Xa en lo que respecta a la catálisis de la protrombina. La trombina generada a partir de esto es la encargada de la activación del factor XIII y del inhibidor de la fibrinólisis activado por trombina (TAFI), sustancias que ayudan a conseguir y mantener la estabilidad del coágulo (11-13).
Se puede afirmar, entonces, que este modelo contempla una vía única y la focalización del proceso en las superficies celulares (14, 15).
Sistemas anticoagulantes: con el fin de evitar la formación de cantidades excesivas de trombina, que son innecesarias y potencialmente perjudiciales, el sistema hemostásico está controlado de forma estricta por los anticoagulantes naturales. Estos compuestos están localizados en el endotelio vascular, de los que se destacan el inhibidor de la vía del factor tisular (TFPI, por su sigla en inglés), la antitrombina y la proteína C (6).
El TFPI ejerce su función al unirse al complejo FT-FVII, con lo que evita que este active los factores IX y X. La antitrombina tiene una acción doble al actuar directamente sobre la trombina pero también sobre los factores IXa y Xa. La proteína C se halla en dos formas: la endotelial, que necesita de la trombomodulina para que actúe como receptor al momento de la activación por la trombina, y la proteína C circulante, que se une a un receptor endotelial.
Estos dos componentes permiten la transformación de proteína C a proteína C activada, que en presencia de su cofactor, la proteína S, inhibe los factores V y VIII (16, 17).
HEMOSTASIS PRIMARIA
Aproximadamente un tercio de los pacientes con enfermedad hepática crónica desarrolla trombocitopenia, la cual empeora a medida que progresa la enfermedad (18). En pacientes con cirrosis avanzada, hasta el 90% de las plaquetas pueden ser almacenadas en el bazo; sin embargo, en estos pacientes el recuento de plaquetas periféricas, por lo general, permanece solo moderadamente disminuido (19). Otros mecanismos que están implicados en este fenómeno son la producción disminuida de trombopoyetina por el hígado y la vida media plaquetaria reducida, relacionada con la presencia de anticuerpos contra las glicoproteína (GP) IIb-IIIa y GP Ib/I producidos por los linfocitos B, especialmente en las cirrosis causadas por el virus de la hepatitis B (VHB), el virus de la hepatitis C (VHC), la cirrosis biliar primaria y la colangitis esclerosante primaria (20-23).
En los pacientes con cirrosis por alcohol, la producción de las plaquetas está alterada por el déficit de ácido fólico y por los efectos tóxicos del etanol en la megacariopoyesis (24). El VHC también está asociado con un efecto mielosupresor que podría sumarse a las lesiones mencionadas previamente (22). Las alteraciones plaquetarias no se limitan exclusivamente a la disminución en su número, también pueden encontrarse algunos cambios funcionales como la menor producción de tromboxano A2, defectos en la GPIb (25), agregación alterada en respuesta al difosfato de adenosina (ADP), ácido araquidónico, colágeno y trombina, lo que refleja probablemente mecanismos alterados de transducción de señales y disminución en el número de receptores plaquetarios funcionales como consecuencia de la proteólisis por la plasmina (26, 27). Adicional a esto, la producción excesiva por parte del endotelio de algunos inhibidores de la agregación plaquetaria como el óxido nítrico y las prostaciclinas, además de la proteólisis plaquetaria por la plasmina, contribuyen a la alteración en la activación de estas células (28).
Por su parte, en la hemostasis primaria del paciente cirrótico, el factor de von Willebrand se encuentra elevado de tal manera que puede alcanzar hasta 10 veces el valor normal y actuar como un elemento compensador para las alteraciones plaquetarias en número y función (19, 29).
HEMOSTASIS SECUNDARIA
Como se mencionó previamente, el hígado es el órgano donde se sintetizan la mayoría de los factores de coagulación, por lo que se puede encontrar una alteración numérica o funcional de estos en enfermedades hepáticas crónicas (30).
Los factores de la coagulación dependientes de la vitamina K, la protrombina y los factores VII, IX y X pueden estar alterados tanto cuantitativa como cualitativamente a consecuencia de la disminución de la γ-carboxilación, la cual afecta la conversión de residuos de ácido glutámico a ácido γ-carboxiglutámico en los precursores proteicos (31). La reducción de esta actividad enzimática se debe a la deficiencia de vitamina K o a la alteración en la síntesis de la carboxilasa dependiente de vitamina K en el hígado (32). Cuando el proceso de γ-carboxilación falla, se sintetizan proteínas inertes denominadas proteínas inducidas por ausencia de vitamina K (PIVKA, por su sigla en inglés), que no pueden ser ligadas por los puentes de calcio (18, 31).
El fibrinógeno es un reactante de fase aguda y permanece en niveles normales, e incluso elevados, en pacientes con enfermedad hepática leve a moderada, mientras que los niveles disminuidos relacionados con una menor síntesis son vistos únicamente en enfermedad crónica severa o falla hepática aguda (19). A pesar de estas altas concentraciones, entre el 60% y el 70% de este compuesto es no funcional ya que tiene un contenido excesivo de ácido siálico y cadenas α anormales, lo que altera su polimerización y hace que este aumento en la cantidad no resulte en la mayor formación de fibrina (33).
ALTERACIONES EN EL SISTEMA FIBRINOLITICO
El sistema fibrinolítico se encarga de la degradación de la fibrina y se activa cuando hay depósito de esta en algún lugar del sistema vascular; así ejerce un control estricto sobre el sistema de la coagulación. La conversión de plasminógeno en plasmina es un proceso en el que intervienen diferentes activadores, como el activador tisular del plasminógeno (tPA) y el activador tipo uroquinasa del plasminógeno (uPA). Estos son contrarrestados por inhibidores de la activación como el inhibidor del activador del plasminógeno (PAI-1), el inhibidor α1 de la plasmina y el más recientemente descrito, inhibidor de la fibrinólisis activado por trombina (TAFI). Este funciona para remover los residuos de lisina de la fibrina y prevenir así la unión y activación de plasminógeno en su superficie (34).
Todas las proteínas implicadas en este proceso, a excepción del tPA y PAI-1, son sintetizadas en el hígado (18). La cirrosis, al igual que otras enfermedades hepáticas crónicas, está asociada con bajos niveles de plasminógeno, α2 antiplasmina, inhibidor de plasmina, factor XIII y TAFI, pero con concentraciones aumentadas de PAI-1 y tPA, éste último debido a la disminución de su eliminación y al aumento de la liberación por el endotelio activado (5, 35).
El balance del sistema fibrinolítico es de gran importancia para evitar la generación no deseada de plasmina; es así como la perturbación de dicho balance puede resultar en hiper o hipofibrinólisis, que pueden asociarse con eventos hemorrágicos o trombóticos, respectivamente (5). De manera clásica, en los pacientes cirróticos se ha descrito un estado de hiperfibrinólisis; sin embargo, la evidencia de dicho estado aún no se valida del todo ya que esta afirmación se basa en las mediciones de componentes individuales más que en pruebas globales, lo que no brinda una imagen completa del complejo balance fibrinolítico, regulado de manera estrecha por los activadores y antiactivadores (25). Últimamente se le ha prestado mucha atención al papel que el TAFI desempeña; se ha postulado que bajos niveles de este inhibidor podrían explicar la hiperfibrinólisis en los pacientes cirróticos. Lisman evaluó dicha hipótesis a través de la medición de componentes individuales y con el uso de pruebas globales que estimaban la capacidad fibrinolítica total. Concluyó que la deficiencia de TAFI en cirróticos no está asociada con hiperfibrinólisis; en cambio sugirió que al igual que en la coagulación, en este sistema se logra un nuevo balance entre los factores pro y antifibrinolíticos (36). A pesar de esto, la controversia no ha sido resuelta debido a que otros autores han reportado resultados opuestos de una relación entre TAFI e hiperfibrinólisis (37).
HIPERCOAGULACION EN ENFERMEDAD HEPATICA
La deficiencia de factores procoagulantes en enfermedad hepática crónica pudiese ser la responsable de la tendencia al sangrado de los pacientes con cirrosis. No obstante, también se ha demostrado una disminución en la producción de anticoagulantes endógenos como la proteína C, proteína S, trombomodulina y tPA (38, 39). Además, en la cirrosis se presenta una reducción en la producción proteica, que se traduce en una reserva disminuida de factores hacia ambos lados del sistema, lo que a su vez reduce la capacidad de compensar incluso pequeñas variaciones en la hemostasis sanguínea, que en ocasiones favorece un estado de hipercoagulabilidad. Un ejemplo sería la disminución en las reservas de proteína C: pequeñas deficiencias de esta generan alteración del balance hemostásico y favorecen el estado protrombótico (4).
En la cirrosis se presenta un estado constante de vasodilatación sistémica. Este afecta especialmente el lecho esplácnico, situación que contribuye al desarrollo de estasis vascular, la cual se acompaña de altas concentraciones de factores procoagulantes. De esta manera aporta a la alta prevalencia de trombosis portal (40).
Como manifestaciones clínicas del predominio del estado de hipercoagulabilidad se encuentra la enfermedad trombótica macrovascular, que puede presentarse como tromboembolismo pulmonar (TEP), trombosis venosa profunda (TVP) o trombosis de la vena porta. Debido a que en la práctica clínica se le da mayor importancia a la detección de las anormalidades en la coagulación que favorezcan el sangrado, este fenómeno muy pocas veces se diagnostica antes de que se presenten dichos desenlaces (4).
Otra manifestación es la trombosis microvascular, cuyas principales características son la hipertensión portal y portopulmonar (41). Posiblemente su fisiopatología se explica por la pérdida de la barrera endotelial, lo que activa la cascada de la coagulación y expone el músculo liso a señales de vasoconstricción, proliferación y trombosis, con el posterior desarrollo de arteriopatía (42). La presencia de microtrombos en la microvasculatura hepática se ha asociado con mayor inflamación hepática, fibrosis y rápido establecimiento de cirrosis, fenómeno que hace parte del proceso denominado extinción de parénquima (43).
"REBALANCE" HEMOSTASICO EN CIRROSIS
Los pacientes con enfermedad hepática crónica presentan alteraciones de la coagulación, tanto de las vías procoagulantes como anticoagulantes (4). Aunque las pruebas hemostásicas rutinarias como el recuento de plaquetas, tiempo de protrombina (TP), tiempo de tromboplastina parcial activado (TTPa) e INR pueden indicar una tendencia hacia el sangrado, diferentes estudios han demostrado que sus valores no se asocian con ni predicen el riesgo de presentar trombosis o hemorragia, con lo que se demuestra poca efectividad y correlación clínica, especialmente si se toma en cuenta que los exámenes de laboratorio actuales no brindan una mirada integral al sistema de coagulación (44).
En pacientes con cirrosis, el sistema de hemostasis se encuentra en un estado de "rebalance", ya que los cambios en alguna de estas vías generan, a su vez, cambios compensatorios en la otra (véase tabla 1) (19, 34, 45). Es frecuente observar trombocitopenia y defectos en la función de las plaquetas; sin embargo, estos problemas son compensados por los niveles elevados de factor de von Willebrand y la disminución en los niveles de la proteasa encargada de la degradación de este factor (19, 29, 46). El resultado de esto es una mejora en la adhesión plaquetaria, evidenciado en las pruebas in vitro (44).
Tabla 1. Alteraciones en el sistema hemostásico en pacientes con enfermedad hepática.

Se ha demostrado que los pacientes con cirrosis tienen intacta la capacidad para producir trombina (47). Adicional a esto, existe en los pacientes con enfermedad hepática crónica una resistencia intrínseca a la acción de la trombomodulina, el activador fisiológico de la proteína C (48). Finalmente, los niveles reducidos de los inhibidores de la fibrinólisis están balanceados, al menos de manera parcial, con los niveles disminuidos de factores profibrinolíticos, en particular el plasminógeno (36).
El efecto de todos estos cambios es un sistema hemostásico con un nuevo balance, pero aún funcional. No obstante, este balance es más precario e inestable comparado con el de un individuo sano, de tal manera que es posible que pequeños cambios en alguno de sus componentes puedan alterarlo; esto podría explicar la presentación de complicaciones como sangrado o trombosis (44).
PRUEBAS DE LABORATORIO
A pesar de su uso ampliamente difundido, se ha demostrado que las pruebas utilizadas para evaluar la hemostasis primaria y secundaria en pacientes con cirrosis no son de utilidad en la práctica clínica por su poco valor como predictores de riesgo de sangrado, particularmente por que estas se afectan cuando los niveles de factores procoagulantes se encuentran entre un 30%-40% de sus valores normales (49-52).
El tiempo de sangría, considerado de larga data como el examen adecuado para la evaluación de la hemostasis primaria, es prolongado hasta en el 40% de los pacientes con cirrosis; sin embargo, la relevancia clínica de dicho hallazgo no ha sido claramente establecida. Estudios que evaluaron el papel de la desmopresina en el tratamiento estándar de pacientes con cirrosis encontraron que a pesar de que dicho fármaco disminuyó el tiempo de sangría, no logró reducir el sangrado en pacientes que sometidos a procedimientos de alto riesgo como la hepatectomía; tampoco disminuyó la tasa de hemorragia variceal, lo que indica que la corrección del tiempo de sangría no resulta necesariamente en mejoría de la hemostasis primaria (53, 54).
El TP evalúa la vía intrínseca de la coagulación detectando deficiencias de factores de coagulación II, V, VII, X y fibrinógeno, mientras que el TTPa evalúa la vía extrínseca. Esta prueba identifica la deficiencia de todos los factores de la coagulación excepto los factores VII y XIII. El TP y el TTPa no reflejan de manera adecuada el nuevo balance en la coagulación que se desarrolla en los pacientes con enfermedad hepática, porque estos solo miden la fase procoagulante y en las hepatopatías crónicas también hay una disminución de los anticoagulantes naturales (47). Para que ejerza toda su función anticoagulante, la proteína C debe ser activada por la trombina y que este proceso sea aumentado por la trombomodulina, glicoproteína que se encuentra en las células endoteliales. A su vez, la trombomodulina necesita ser activada por glucosaminoglucanos endoteliales como el heparán-sulfato (55). En este punto cabe anotar que ni el plasma ni los reactivos utilizados para la realización del TP o TTPa contienen trombomodulina o glucosaminoglucanos, motivo por el cual se puede afirmar que estas pruebas representan principalmente la trombina generada por los factores procoagulantes, pero en menor medida la inhibición ejercida por los anticoagulantes naturales (47).
A pesar de que en los pacientes con cirrosis las pruebas como el TP y el TTPa permanecen alteradas, cuando el balance entre factores pro y anticoagulantes es medido por otros métodos que evalúan de forma más completa el funcionamiento del sistema de la coagulación, este resulta ser normal. Es así como Tripodi y colaboradores demostraron que la formación de trombina en los pacientes cirróticos es normal o no difiere significativamente de la producida por personas sin alteración hepática cuando el recuento plaquetario está entre 50 000 y 60 000 plaquetas por mililitro, de modo que su producción es óptima cuando el recuento es igual o mayor a 100 000 plaquetas por mililitro (56). Por lo tanto, se puede inferir que la coagulación en pacientes con cirrosis es normal si los niveles de plaquetas son suficientemente altos para sostener la generación normal de trombina; de esta manera se recomienda que al momento de realizar procedimientos que generen riesgo moderado de sangrado en estos pacientes el recuento plaquetario mínimo sea de 50 000/mL, mientras que para procedimientos de riesgo alto este debe estar cerca de los 100 000/mL (57).
También se ha demostrado que bajo condiciones fisiológicas de flujo, las plaquetas de pacientes con cirrosis son capaces de interactuar normalmente con el colágeno y el fibrinógeno, siempre y cuando se ajuste el recuento plaquetario y el hematocrito a niveles normales; por ende in vivo, los defectos de nú0mero y función plaquetaria parecen no ser tan relevantes como se ha considerado hasta ahora (36, 56, 58).
Inicialmente el INR fue validado para la estandarización del TP únicamente en pacientes que reciben terapia con antagonistas de vitamina K como la warfarina (59). Sin embargo, también se usa en la evaluación de los pacientes con enfermedad hepática, e incluso hace parte del modelo para la predicción de supervivencia en pacientes con enfermedad hepática en estado terminal (MELD, por su sigla en inglés) (60, 61). El problema radica en el uso del índice internacional de sensibilidad (ISI) para el cálculo del INR, que genera una variabilidad interlaboratorio entre el 25% y el 78% en la media del INR de pacientes con enfermedad hepática (62-66). Adicionalmente el ISI no está validado en pacientes cirróticos en quienes la coagulopatía es más compleja, ya que además de compartir las deficiencias de factores de la coagulación que presentan los pacientes anticoagulados, también tienen deficiencias de factor V y de síntesis del fibrinógeno (67).
Dicho lo anterior, es más apropiado pensar que la historia de sangrados previos, la presencia y el tamaño de las várices esofágicas, el grado de hipertensión portal y las comorbilidades tales como la falla renal o anemia, tienen un mayor valor predictivo de sangrado en pacientes cirróticos que la alteración de los exámenes de coagulación rutinarios (31).
NUEVAS PRUEBAS
El analizador de función plaquetaria (FPA, por su sigla en inglés) es una prueba (o test) rápida e in vitro que permite la medición cuantitativa de la hemostasis primaria. Este dispositivo funciona con sangre que fluye constantemente en un sistema vacumm a través de un capilar y una apertura microscópica recubierta por colágeno y agonistas. El tiempo que demore en cerrar la apertura es un indicador de adhesión y agregación plaquetaria (68, 69).
Tripodi y colaboradores crearon la prueba de generación de trombina que, como su nombre lo indica, está diseñada para investigar la generación de esta peptidasa en presencia de inhibidores de la coagulación (47). En dicha prueba se activa la coagulación con pequeñas cantidades de factor tisular como desencadenante y fosfolípidos como sustitutos de plaquetas (70, 71). Mediante esta prueba se obtiene la curva de generación de trombina, que es una gráfica de su concentración contra el tiempo, en la que se determina el potencial endógeno de este factor, que es la cantidad de trombina que una muestra de plasma puede generar bajo las condiciones de la prueba y que representa el balance entre factores procoagulantes y anticoagulantes. Es la prueba que más se acerca a las condiciones in vivo, aunque requiere de mayor investigación y validación (72).
DISCUSION
Contrario al paradigma por el que la cirrosis se consideraba una enfermedad en la que la tendencia de los pacientes era principalmente hacia el sangrado, actualmente se ha demostrado que coexiste un estado de hipercoagulabilidad que se manifiesta con desenlaces trombóticos micro o macrovasculares pocas veces atribuidos a esta condición. Paralelo a estos hallazgos se postula que el sistema de coagulación en los pacientes con cirrosis se encuentra en un estado de "rebalance" que, si bien es más precario que en las personas sanas, les permite funcionar adecuadamente aun en situaciones de estrés significativo siempre y cuando se ajuste el recuento plaquetario y el hematocrito a los niveles apropiados para su condición. Como las pruebas comúnmente utilizadas para la evaluación del estado de coagulación de estos pacientes no muestran este nuevo rebalance ya que se centran en las alteraciones del componente procoagulante, no deben considerarse herramientas útiles para la predicción del riesgo de sangrado o de complicaciones trombóticas; por ende, no es apropiada la toma de decisiones con base en la alteración de sus resultados.
REFERENCIAS
1. Basili S, Raparelli V, Violi F. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? Yes. Eur J Intern Med 2010; 21(2): 62-4. [ Links ]
2. Tripodi A, Primignani M, Mannucci PM. Abnormalities of hemostasis and bleeding in chronic liver disease: the paradigm is challenged. Intern Emerg Med 2010; 5(1): 7-12. [ Links ]
3. Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost 2004; 91(2): 267-75. [ Links ]
4. Northup PG. Hypercoagulation in liver disease. Clin Liver Dis 2009; 13(1): 109-16. [ Links ]
5. Tripodi A. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? No. Eur J Intern Med 2010; 21(2): 65-9. [ Links ]
6. Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost 2006; 32(Suppl 1): 32-8. [ Links ]
7. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol 2007; 27(8): 1687-93. [ Links ]
8. Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci 2006; 11: 1349-59. [ Links ]
9. Monroe DM, Hoffman M, Roberts HR. Transmission of a procoagulant signal from tissue factor-bearing cell to platelets. Blood Coagul Fibrinolysis 1996; 7(4): 459-64. [ Links ]
10. Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol 1999; 19(1):170-7. [ Links ]
11. Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost 2003; 1(7): 1504-14. [ Links ]
12. Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006; 26(1): 41-8. [ Links ]
13. Crawley JT, Zanardelli S, Chion CK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemost 2007; 5(Suppl 1): 95-101. [ Links ]
14. Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost 2001; 38(4 Suppl 12): 958-65. [ Links ]
15. Hoffman MM, Monroe DM. Rethinking the coagulation cascade. Curr Hematol Rep 2005; 4(5): 391-6. [ Links ]
16. Esmon CT. The protein C pathway. Chest 2003; 124(3 Suppl): 26S-32S. [ Links ]
17. Dahlbäck B, Stenflo J. Regulatory mechanisms in hemostasis: natural anticoagulants. En: Hoffman R, Furie B, Benz Jr. EJ, McGlave P, Silberstein LE, Shattil SJ, editors. Hematology: basic principles and practice. 5 edition. Philadelphia: Churchill, Livingstone: Elsevier; 2009. p. 1843-9. [ Links ]
18. New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol 2006; 12(48): 7725-36. [ Links ]
19. Lisman T, Leebeek FW. Hemostatic alterations in liver disease: a review on pathophysiology, clinical consequences, and treatment. Dig Surg 2007 24(4): 250-8. [ Links ]
20. Goulis J, Chau TN, Jordan S, Mehta AB, Watkinson A, Rolles K, et al. Thrombopoietin concentrations are low in patients with cirrhosis and thrombocytopenia and are restored after orthotopic liver transplantation. Gut 1999; 44(5): 754-8. [ Links ]
21. Kajihara M, Kato S, Okazaki Y, Kawakami Y, Ishii H, Ikeda Y, et al. A role of autoantibody-mediated platelet destruction in thrombocytopenia in patients with cirrhosis. Hepatology 2003; 37(6): 1267-76. [ Links ]
22. Nagamine T, Ohtuka T, Takehara K, Arai T, Takagi H, Mori M. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol 1996; 24(2): 135-40. [ Links ]
23. Feistauer SM, Penner E, Mayr WR, Panzer S. Target platelet antigens of autoantibodies in patients with primary biliary cirrhosis. Hepatology 1997; 25(6): 1343-5. [ Links ]
24. Levine RF, Spivak JL, Meagher RC, Sieber F. Effect of ethanol on thrombopoiesis. Br J Haematol 1986; 62(2): 345-54. [ Links ]
25. Tripodi A. Hemostasis abnormalities in liver cirrhosis: myth or reality? Pol Arch Med Wewn 2008; 118(7-8): 445-8. [ Links ]
26. Escolar G, Cases A, Vinas M, Pino M, Calls J, Cirera I, et al. Evaluation of acquired platelet dysfunctions in uremic and cirrhotic patients using the platelet function analyzer (PFA-100 ): influence of hematocrit elevation. Haematologica 1999; 84(7): 614-9. [ Links ]
27. Pasche B, Ouimet H, Francis S, Loscalzo J. Structural changes in platelet glycoprotein IIb/IIIa by plasmin: determinants and functional consequences. Blood 1994; 83(2): 404-14. [ Links ]
28. Cahill PA, Redmond EM, Sitzmann JV. Endothelial dysfunction in cirrhosis and portal hypertension. Pharmacol Ther 2001; 89(3): 273-93. [ Links ]
29. Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006; 44(1): 53-61. [ Links ]
30. Kerr R. New insights into haemostasis in liver failure. Blood Coagul Fibrinolysis 2003; 14(Suppl 1): S43-5. [ Links ]
31. Pluta A, Gutkowski K, Hartleb M. Coagulopathy in liver diseases. Adv Med Sci 2010; 55(1): 16-21. [ Links ]
32. Blanchard RA, Furie BC, Jorgensen M, Kruger SF, Furie B. Acquired vitamin K-dependent carboxylation deficiency in liver disease. N Engl J Med 1981; 305(5): 242-8. [ Links ]
33. Francis JL, Armstrong DJ. Fibrinogen-bound sialic acid levels in the dysfibrinogenaemia of liver disease. Haemostasis 1982; 11(4): 215-22. [ Links ]
34. Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol 2007; 46(4): 727-33. [ Links ]
35. Leiper K, Croll A, Booth NA, Moore NR, Sinclair T, Bennett B. Tissue plasminogen activator, plasminogen activator inhibitors, and activator-inhibitor complex in liver disease. J Clin Pathol 1994; 47(3): 214-7. [ Links ]
36. Lisman T, Leebeek FW, Mosnier LO, Bouma BN, Meijers JC, Janssen HL, et al. Thrombin-activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology 2001; 121(1): 131-9. [ Links ]
37. Colucci M, Binetti BM, Branca MG, Clerici C, Morelli A, Semeraro N, et al. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology 2003; 38(1): 230-7. [ Links ]
38. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol 2002; 37(2): 280-7. [ Links ]
39. Vukovich T, Teufelsbauer H, Fritzer M, Kreuzer S, Knoflach P. Hemostasis activation in patients with liver cirrhosis. Thromb Res 1995; 77(3): 271-8. [ Links ]
40. Artiko V, Obradovic V, Petrovic M, Perisic M, Stojkovic M, Sobic-Saranovic D, et al. Hepatic radionuclide angiography and Doppler ultrasonography in the detection and assessment of vascular disturbances in the portal system. Hepatogastroenterology 2007; 54(75): 892-7. [ Links ]
41. Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl 2005; 11(9): 1107-11. [ Links ]
42. Fallon MB. Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary syndrome. J Clin Gastroenterol 2005; 39(Suppl 2): S138-42. [ Links ]
43. Anstee QM, Wright M, Goldin R, Thursz MR. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis 2009; 13(1): 117-26. [ Links ]
44. Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol 2010; 53(2): 362-71. [ Links ]
45. Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, Reddy KR, et al. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology 2006; 44(4): 1039-46. [ Links ]
46. Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood 2001; 98(9): 2730-5. [ Links ]
47. Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005; 41(3): 553-8. [ Links ]
48. Tripodi A, Primignani M, Chantarangkul V, Dell'Era A, Clerici M, de Franchis R, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology 2009; 137(6): 2105-11. [ Links ]
49. Ewe K. Bleeding after liver biopsy does not correlate with indices of peripheral coagulation. Dig Dis Sci 1981; 26(5): 388-93. [ Links ]
50. Segal JB, Dzik WH. Paucity of studies to support that abnormal coagulation test results predict bleeding in the setting of invasive procedures: an evidence-based review. Transfusion 2005; 45(9): 1413-25. [ Links ]
51. Boks AL, Brommer EJ, Schalm SW, Van Vliet HH. Hemostasis and fibrinolysis in severe liver failure and their relation to hemorrhage. Hepatology 1986; 6(1): 79-86. [ Links ]
52. Tellez-Avila FI, Chavez-Tapia NC, Torre-Delgadillo A. [Coagulation disorders in cirrhosis]. Rev Invest Clin 2007; 59(2): 153-60. [ Links ]
53. Pivalizza EG, Warters RD, Gebhard R. Desmopressin before liver transplantation. Can J Anaesth 2003; 50(7): 748-9. [ Links ]
54. Wong AY, Irwin MG, Hui TW, Fung SK, Fan ST, Ma ES. Desmopressin does not decrease blood loss and transfusion requirements in patients undergoing hepatectomy. Can J Anaesth 2003; 50(1): 14-20. [ Links ]
55. Dahlbäck B. Progress in the understanding of the protein C anticoagulant pathway. Int J Hematol 2004; 79(2): 109-16. [ Links ]
56. Tripodi A, Primignani M, Chantarangkul V, Clerici M, Dell'Era A, Fabris F, et al. Thrombin generation in patients with cirrhosis: the role of platelets. Hepatology 2006; 44(2): 440-5. [ Links ]
57. Holland LL, Brooks JP. Toward rational fresh frozen plasma transfusion: The effect of plasma transfusion on coagulation test results. Am J Clin Pathol 2006; 26(1): 133-9. [ Links ]
58. Lisman T, Adelmeijer J, de Groot PG, Janssen HL, Leebeek FW. No evidence for an intrinsic platelet defect in patients with liver cirrhosis--studies under flow conditions. J Thromb Haemost 2006; 4(9): 2070-2. [ Links ]
59. Kirkwood TB. Calibration of reference thromboplastins and standardisation of the prothrombin time ratio. Thromb Haemost 1983; 49(3): 238-44. [ Links ]
60. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33(2): 464-70. [ Links ]
61. Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KV, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2): 353-8. [ Links ]
62. Arjal R, Trotter JF. International normalized ratio of prothrombin time in the model for end-stage liver disease score: an unreliable measure. Clin Liver Dis 2009; 13(1): 67-71. [ Links ]
63. Tripodi A, Chantarangkul V, Primignani M, Fabris F, Dell'Era A, Sei C, et al. The international normalized ratio calibrated for cirrhosis (INR(liver)) normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology 2007; 46(2): 520-7. [ Links ]
64. Kovacs MJ, Wong A, MacKinnon K, Weir K, Keeney M, Boyle E, et al. Assessment of the validity of the INR system for patients with liver impairment. Thromb Haemost 1994; 71(6): 727-30. [ Links ]
65. Denson KW, Reed SV, Haddon ME, Woodhams B, Brucato C, Ruiz J. Comparative studies of rabbit and human recombinant tissue factor reagents. Thromb Res 1999; 94(4): 255-61. [ Links ]
66. Robert A, Chazouillères O. Prothrombin time in liver failure: time, ratio, activity percentage, or international normalized ratio? Hepatology 1996; 24(6): 1392-4. [ Links ]
67. Kamath PS, Kim WR. The international normalized ratio of prothrombin time in the model for end-stage liver disease score: a reliable measure. Clin Liver Dis 2009; 13(1): 63-6. [ Links ]
68. Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apher Sci 2003; 28(3): 307-17. [ Links ]
69. Kundu SK, Heilmann EJ, Sio R, Garcia C, Davidson RM, Ostgaard RA. Description of an in vitro platelet function analyzer--PFA-100. Semin Thromb Hemost 1995; 21(Suppl 2): 106-12. [ Links ]
70. Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 2003; 33(1): 4-15. [ Links ]
71. Chantarangkul V, Clerici M, Bressi C, Giesen PL, Tripodi A. Thrombin generation assessed as endogenous thrombin potential in patients with hyper- or hypo-coagulability. Haematologica 2003; 88(5): 547-54. [ Links ]
72. Tripodi A. Tests of coagulation in liver disease. Clin Liver Dis 2009; 13(1): 55-61. [ Links ]

