










Services on Demand
Journal
Article
 text in
text in  Spanish (pdf)
Spanish (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957On-line version ISSN 2500-7440
Rev Col Gastroenterol vol.27 no.2 Bogotá Apr./June 2012
Old and new paradigms for cirrhosis associated coagulation abnormalities
Yesid Alberto Saavedra González, Est. (1), Laura Margarita Ovadía Cardona, Est. (1), Octavio Germán Muñoz Maya, MD. (2), Gonzalo Correa Arango, MD. (2)
(1) 10th Semester Student in the Gastro-hepatology Group in the Faculty of Medicine at the Universidad de Antioquia in Medellín, Colombia.
(2) Medical Internist specializing in Hepato-pathology in the Gastro-hepatology Group at the Hospital Pablo Tobón Uribe and the Universidad de Antioquia in Medellín, Colombia.
Received: 06-12-11 Accepted: 15-05-12
Abstract
In liver disease one of the liver's functions related to blood coagulation, hemostasis, is disturbed. This manifests as hypercoagulability in some patients and as excessive bleeding in others.
Objective: The objective of this study was to review current evidence about blood coagulation disorders in cirrhotic patients.
Methods: We conducted a search using the following "MESH" terms: blood coagulation disorders, cirrhosis, hemostasis, hypercoagulability, bleeding and blood coagulation tests. From the results articles whose titles and abstracts best fit with the aim of this review were chosen.
Results: From 146 articles obtained, 76 were selected and used to construct this article.
Conclusion: Despite the diverse blood coagulation disorders present in cirrhotic patients, the hemostatic system accomplishes a new balance that currently used blood coagulation tests fail to show. This is the why these cannot be considered useful tools for predicting the risk of bleeding or thrombotic complications.
Key words
Blood coagulation disorders, cirrhosis, hypercoagulability, bleeding, blood coagulation tests.
INTRODUCTION
The liver performs several roles in both primary and secondary hemostasis (1). It is the place where fibrinogen and coagulation factors II, V, VII, IX, X, XI, XII and XIII (2) are synthesized. Factor VIII is synthesized primarily by sinusoidal endothelial cells and to a lesser extent by endothelial cells of the lung, the kidney, the spleen and the brain (3).
The coagulation system consists of a complex balance between procoagulant components, anticoagulant components and the fibrinolytic system. This system functions during the hemostatic process and offers a very quick response to endothelial injuries with fibrin deposits, platelet aggregation and clot formation. In a healthy individual, the events of coagulation are counteracted by anticoagulant activity which prevents the inappropriate extension of the clot, minimizes local ischemia and promotes lysis once hemostasis has been reached. In the context of liver disease this balance is altered resulting in the dominance of one of the two components. This can manifest as hypercoagulation in some patients and as excessive bleeding in others (4).
MATERIALS AND METHODS
Pubmed and Scielo databases were searched for review and original articles on cirrhosis and clotting disorders. Search terms were "MESH": Blood coagulation disorders; cirrhosis; hemostasis; hypercoagulability; bleeding; blood coagulation tests. From the results found only the articles whose titles and abstracts best suited the purpose of this review were chosen. Of the 146 abstracts read, 72 were chosen to be read completely. The information gathered from these articles was complemented with articles referenced by articles on our primary list. This paper was constructed from all of these sources.
RESULTS
A new model of coagulation
The traditional coagulation model is an enzyme cascade consisting of series of sequential steps in which the activation of a coagulation factor activates the next factor. The ultimate purpose is production of fibrin the structural component in coagulation (5). This is the well known model of the intrinsic and extrinsic pathways of coagulation. Despite its long acceptance, recent studies describing the fundamental role of the cellular component have made it well understood that hemostasis is not possible without platelets. In addition, it has been proven that different types of cells express procoagulant proteins, anticoagulants and receivers for multiple components of hemostasis. This has meant a new paradigm to explain the reactions that take place during the hemostatic process (6).
According to the current vision, coagulation is produced in three interrelated stages (Figure 1).
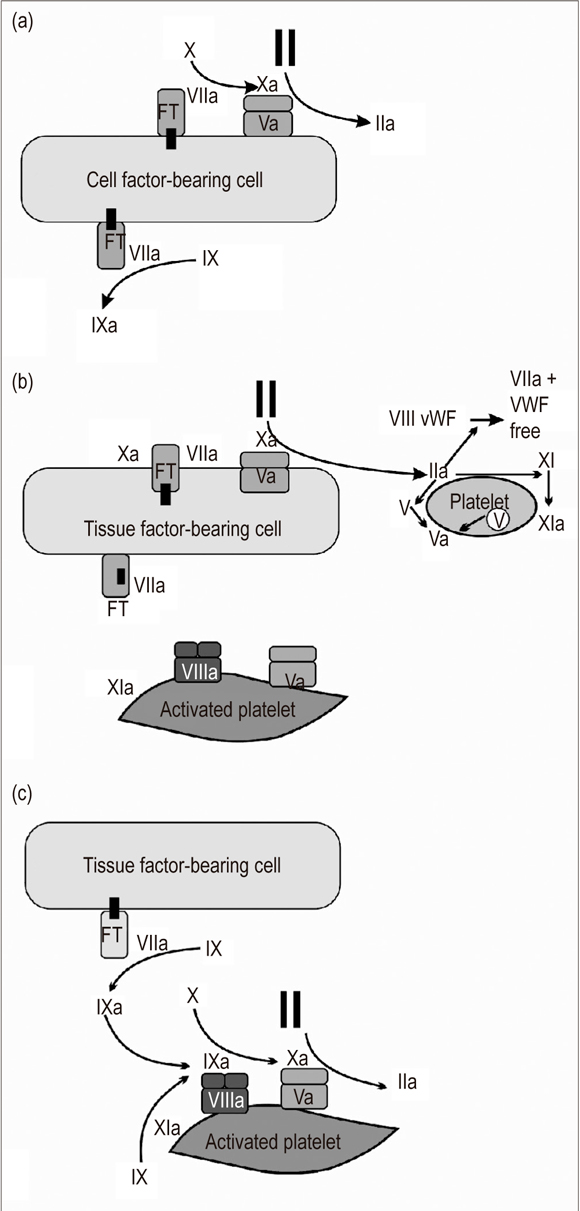
Figure 1. Cellular Model of Coagulation: (a) Initiation, (b) Amplification, (c) Propagation. (Modified on the basis of chart taken from Hoffman M. A cell-based model of coagulation and the role of factor VIIa. Blood Rev. 2003; 17 Suppl 1: S1 5).
Phase 1. Initiation: The tissue factor (TF), an integral component of the cellular membrane, is the primary starter for in vivo coagulation. It expresses numerous cellular types including circulating monocytes, neutrophils, fibroblasts and endothelial cells (7). When a vascular injury occurs and blood makes contact with the subendothelium, and an interaction between cellular TF and circulating factor VII is activated. The TF-VIIa complexes then activate IX and X factors. Factor Xa combines with factor Va on the cellular surface resulting in the production of small quantities of thrombin. The thrombin will then participate in the activation of platelets and VIII factor in the next phase (8).
Phase 2. Amplification: The liver injury allows platelets and plasma components to come into contact with extravascular tissues. Once this has happened, the platelets adhere to the subendothelial matrix and activate those sites where the TF is exposed. The thrombin generated in phase 1 amplifies the procoagulant signal by activating factors V, VIII and XI which are located on the platelet surface. They serve as mediators in the reactions that take place during the next phase (9, 10).
Phase 3. Spread: This phase is characterized by two principal phenomena. In the first, the tenase complex (VIIIa factor, IXa factor, calcium (Ca++) and, phospholipids) serves as a catalyst promoting the conversion of Xa factor. In the second, the prothrombinase complex, (Xa factor, Va factor, Ca++, and phospholipids) promotes the generation of large amounts of thrombin which are necessary for the formation of a clot resistant to fibrinolysis. This is what has been called thrombin explosion. The prothrombinase complex is fundamental for the final stage of this phase because it is 300,000 times more active than the Xa factor as a catalyst for prothrombin. The thrombin generated here activates the XIII factor and thrombin activatable fibrinolysis inhibitor (TAFI) substances which help achieve and maintain the stability of the clot (11-13).
We can say that this model contemplates only one pathway and that it is focused on the cellular surfaces (14, 15).
Anticoagulant systems: In order to avoid excessive quantities of thrombin which are unnecessary and potentially detrimental, the hemostatic system is strictly controlled by natural anticoagulants. These compounds are located in the vascular endothelium. The most important of them are tissue factor pathway inhibitors (TFPI), antithrombin and protein C (6).
TFPI exerts its effects by binding to the TF-FVIII complex to prevent activation of IX and X factors. Antithrombin acts directly on thrombin but also acts on IXa and Xa factors. Protein C is found in two forms: endothelial protein C which needs thrombomodulin so it can act as a receptor at the moment of activation by thrombin, and circulating protein C which joins with an endothelial receptor.
Both of these compounds allow the transformation of protein C into activated protein C, which in presence of cofactor protein S, inhibits V and VIII factors (16, 17).
PRIMARY HEMOSTASIS
Approximately one third of all patients with chronic liver disease develop thrombocytopenia which worsens as the disease progresses (18). Among patients with advanced cirrhosis almost 90% of the platelets can be stored in the spleen. These patients usually have a moderately low count of peripheral platelets (19). Other mechanisms implied in this phenomenon are the decrease of thrombopoietin production by the liver (20) and reduced platelet life spans. This is related to the presence of antibodies against glycoprotein (GP) IIb-IIIa and GP Ib/I produced by the B lymphocytes. This is especially common in cirrhosis caused by the Hepatitis B virus (21), Hepatitis C virus (22), primary biliary cirrhosis and primary sclerosing cholangitis (23).
Among patients who have cirrhosis due to alcohol consumption the production of platelets is altered by folic acid deficiency and the toxic effects of ethanol on the megakaryopoiesis (24). HCV has also been associated with a myelosuppressive effect that might add to the injuries mentioned above (22). Platelet abnormalities are not limited exclusively to their decreased numbers, but can also involve changes in functioning such as reduced production of throboxane A2, defects in glycoprotein Ib (25), and impaired aggregation in response to adenosine diphosphate (ADP), arachidonic acid, collagen, and thrombin. This probably reflects altered mechanisms of signal transduction and decreasing numbers of functional platelet receptors resulting from proteolysis by plasmin (26, 27). In addition, excessive production of some platelet aggregation inhibitors such as nitric oxide and prostacyclins in the endothelium combined with proteolysis by plasmin platelets contributes to alterations in the activation of these cells (28).
In primary hemostasis cirrhotic patients have levels of von Willebrand factor which sometimes reach 10 times normal levels. This acts as a compensating factor for the number and functional abnormalities of the platelets (19, 20).
SECONDARY HEMOSTASIS
As mentioned, the liver is the organ where the majority of coagulation factors are synthesized. Consequently, changes in the numbers or functions of these factors can be found in chronic liver diseases (30).
Coagulation factors that depend on vitamin K, prothrombin and factors VII, IX and X may be altered both quantitatively and qualitatively as a result of decreases γ-carboxylation. This affects conversion of glutamic acid residues into >γ-carboxyglutamic acid in protein precursors (31). Reduction of these enzyme activities is due to vitamin K deficiency or to alterations in the synthesis of carboxylase which depends on the vitamin K of the liver (32). When the process of y-carboxylation fails, inert proteins are synthesized. These proteins, which cannot be linked by calcium bridges, are known as proteins induced by vitamin K deficiency (PIVKA) (18, 31).
Fibrinogen is an acute phase reactant which remains at normal, or even higher than normal, levels in patients with mild to moderate liver disease. Reduced levels due to lower synthesis are only seen in patients with severe chronic disease or with acute liver failure (19). Despite concentrations of this compound between 60% and 70%, it is not functional due to high levels of sialic acid and abnormal α chains. Because these factors alter polymerization, increased quantities of fibrinogen do not result in the formation of fibrin (33).
ALTERATIONS IN THE FIBRINOLYTIC SYSTEM
The fibrinolytic system, which degrades fibrin, activates when there fibrin deposits anywhere in the vascular system: it exercises strict control over the coagulation system. Activators such as the tissue plasminogen activator (tPA) and the urokinase-type plasminogen activator (uPA) intervene in the process of converting plasminogen into plasmin but are offset by activation inhibitors such as the plasminogen activation inhibitor (PAI-1), α1 inhibitor of plasmin and the more recently described thrombin-activated fibrinolysis inhibitor (TAFI). TAFI works by removing lysine residues from fibrin which prevents binding and activation of the plasminogen on its surface (34).
With exceptions of tPA and PAI-1All, all the proteins implicated in this process are synthesized in the liver (18). Like other chronic liver diseases, cirrhosis is associated with low levels of plasminogen, α2 antiplasmin, plasmin inhibitor, XIII factor and thrombin activatable fibrinolysis factor (TAFI) but with increased concentrations of PAI-1 (5) and tPA. Increased tPA is due to decreased elimination and increased release by the activated endothelium (35).
The balance of the fibrinolytic system has great importance for avoidance of unwanted plasmin generation. Disturbance of this balance can result in hyperfibrinolysis, which is associated with bleeding, or hypofibrinolysis which is associated with thrombosis (5). Classically, hyperfibrinolysis has been described in cirrhotic patients; however this has not been completely validated since it is based on measurements of individual components rather than on global tests. These measurements provide neither a complete picture of the complex fibrinolytic balance nor information about the activators and anti-activators which closely regulate that system (25). Lately, a great deal of attention has been paid to the role played by TAFI. It has been postulated that low levels of this inhibitor may explain hyperfibrinolysis in cirrhotic patients. Lisman evaluated this hypothesis by measuring both individual and global tests which estimated the total fibrinolytic capacity. He concluded that TAFI deficiency in cirrhotic patients is not associated with hyperfibrinolysis. Instead he suggested that, similar to coagulation, a new balance is achieved between pro and anti fibrinolytic factors in this system (36). Nevertheless, this dispute is not yet resolved because other authors have reported opposite findings regarding the relationship between TAFI and hyperfibrinolysis (37).
HYPERCOAGULATION IN LIVER DISEASE
Although procoagulant factor deficiency in chronic liver disease may be responsible for the tendency of cirrhosis patients to bleed, decreased production of endogenous anticoagulants such as protein C, protein S, thrombomodulin and tPA has been also demonstrated (38, 39). Cirrhosis also reduces protein production resulting in decreased reserve factors on both sides of the system. This decreases the ability to compensate for even small variations in blood hemostasis which might favor hypercoagulability in some circumstances. An example of this could be reduction of protein C reserves since small deficiencies of this substance alter the hemostatic balance and promote thrombosis (4).
Cirrhosis presents a constant state of systemic vasodilation which especially affects the splanchnic bed which contributes to development of vascular stasis. This is accompanied by high concentrations of procoagulant factors which contribute to the high prevalence of portal vein thrombosis (40).
A clinical manifestation of the predominance of hypercoagulability is macrovascular thrombosis which may present as pulmonary thromboembolism, as deep vein thrombosis or as portal vein thrombosis. Since clinical practice gives greater importance to coagulation abnormalities that promote bleeding, this phenomenon is rarely diagnosed before these outcomes develop (4).
Another manifestation is microvascular thrombosis which manifests itself primarily as portal and portal-pulmonary hypertension (41). Its pathophysiology might be explained by the loss of the endothelial barrier which triggers a coagulation cascade and exposes the smooth muscle to vasoconstriction, proliferation and thrombosis signals with subsequent development of arteriopathy (42). The presence of microthrombi in the hepatic microvasculature is associated with parenchymal extinction which consists of hepatic inflammation, fibrosis and the rapid establishment of cirrhosis (43).
HEMOSTATIC "REBALANCE" IN CIRRHOSIS
Patients with chronic liver disease present abnormalities along both procoagulant and anticoagulant pathways (4). Although routine hemostatic tests such as platelet counts, prothrombin time (TP), activated partial thromboplastin time (TTPa) and international normalized ratio (INR) can indicate tendencies to bleed, various studies have demonstrated that their values are not related and do not predict the risk of thrombosis or bleeding. Their efficiency and clinical correlation are poor, especially when the fact that current laboratory tests do not provide a comprehensive look at the coagulation system is taken into account (44).
The hemostasis system of cirrhosis patients is "rebalanced" since changes along one pathway generate compensatory changes in the other pathway (See Table 1) (19, 34, 45). It is common to find thrombocytopenia and defects in platelet functions (19), which have been compensated for by high levels of von Willebrand factor (29) and decreased levels of protease (which degrades this factor) (46). The result of this is an improvement in platelet adhesion as evidenced in in-vitro testing (44).
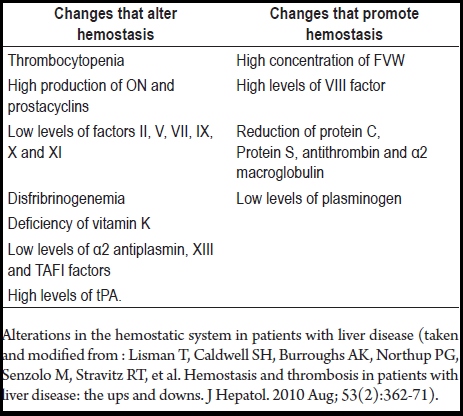
Table 1. Alterations in the hemostatic systems of patients with liver disease.
It has been proven that patients with cirrhosis have intact their capacities for thrombin production (47). In addition, an intrinsic resistance to the action of thrombomodulin, the physiologic activator of protein C, exists in patients with chronic liver disease (48). Finally, reduced levels of fibrinolysis inhibitors are at least partially balanced by reduced levels of profibrinolytic factors, especially plasminogen (36).
The effect of these changes is a hemostatic system with a new balance that is still functional. However, this balance is poorer and less unstable than that of a healthy individual and small changes in any of its components can alter it. This could explain the presentation of complications such as thrombosis and bleeding (44).
Alterations in the hemostatic system in patients with liver disease (taken and modified from : Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol. 2010 Aug; 53(2):362-71).
LABORATORY TESTS
Despite being commonly used, laboratory tests to evaluate primary and secondary hemostasis have been proven to have less use for predicting bleeding in patients with cirrhosis (48, 49, 50, 51). This is particularly true when procoagulant levels are between 30-40% of their normal values (52).
Bleeding time was long considered to be the appropriate test for evaluating primary hemostasis, and it was performed on up to 40% of patients with cirrhosis. Nevertheless, the clinical relevance of its findings has not been established. Studies which evaluate the role of desmopressin in the standard treatment of patients with cirrhosis have found that, despite the fact that it reduces bleeding times, it does not reduce bleeding in patients who undergo high risk procedures such as hepatectomy nor does it reduce the rate of variceal bleeding (53, 54). This indicates that correction of bleeding time does not always result in improvement of primary hemostasis.
TP evaluates the intrinsic pathway of coagulation by detecting deficiencies in coagulation factors II, V, VII, X and fibrinogen while the TTPa evaluates the intrinsic pathway by identifying deficiencies of all coagulation factors except for factors VII and XIII. These tests primarily study the thrombin generated by procoagulant factors, and to a lesser extent the inhibition exerted by natural anticoagulants. These tests only measure the procoagulant phase, but in chronic liver diseases there is also a reduction of natural anticoagulants (47). Protein C must be activated by thrombin to exercise all its anticoagulant functions, and this process is enhanced by thrombomodulin glycoprotein that is found in endothelial cells. In turn, thrombomodulin needs to be activated by endothelial glycosaminoglycans (47). Consequently, TP and the TTPa do not adequately measure the new coagulation balance that develops in patients with liver disease.
Although tests like PT and aPTT show altered balances between pro-and anticoagulant factors in patients with cirrhosis, when this balance is measured by other methods that evaluate it the functioning of the coagulation system more fully, the balance turns out to be normal. Tripodi and his collaborators demonstrated that the formation of thrombin in patients with cirrhosis is normal, and it does no differ significantly from people without liver disorders, when the platelet count is between 50,000 and 60,000 platelets per milliliter even though optimal production is 100,000 platelets or more per milliliter (56). We can infer that patients with cirrhosis will have normal coagulation balances when their platelet levels are high enough to sustain normal generation of thrombin. Consequently, it is recommended that the minimum platelet count for performing a procedure with a moderate risk of bleeding on a patient with cirrhosis should be 50,000/mL while for high risk procedures it should be almost 100,000/mL (57).
It has also been demonstrated that, as long as the platelet and hematocrit counts are at normal levels, under physiological fluid conditions platelets in patients with cirrhosis are capable of normal interaction with collagen and fibrinogen (58). The result is that in vivo the number and function of the platelets are not as relevant as had been considered in the past (36, 56).
Initially the INR was validated for standardization of TP only for patients receiving treatment with vitamin K antagonists such as warfarin (59). However, it is also used for evaluating patients with liver disease and is even used for Model for End-Stage Liver Disease (MELD) scores for survival prediction in patients with terminal liver disease (60, 61). The problem here is that the use of the international sensitivity index (ISI) to calculate INR (62, 63) generates variations in the mean INR of patients with liver disease among laboratories of 25% to 78% (64-66). In addition, the ISI has not been validated for cirrhotic patients who have coagulopathies which are more complex than those of other people. In addition to coagulation factor deficiencies, which patients with poor coagulation also have, patients with cirrhosis also have factor V and fibrinogen synthesis deficiencies (67).
Having said all this, it is more appropriate to think about a patient's previous history of bleeding, the presence and size of esophageal varices, the level of portal hypertension and comorbidities such as renal leakage or anemia, since these are better predictors of bleeding than are alterations in routine coagulation examinations (31).
NEW TESTS
The function platelet analyzer (FPA) is a quick in vitro test that allows quantitative measurement of primary hemostasis. In this device, blood flows constantly into a vacuum system through a capillary and a microscopic opening coated with collagen and agonists. The time it takes for the opening to close is an indicator of platelet adhesion and aggregation (68, 69).
Tripodi and colleagues created the thrombin generation test to study generation of thrombin in the presence of coagulation inhibitors (47). In this test coagulation is activated with small amounts of tissular factor as a trigger and with phospholipids as platelet substitutes (70, 71). This test produces a thrombin generation curve, a graph of time against thrombin concentrations, which determines the endogenous potential of the thrombin. This potential, which is the quantity of thrombin that can be generated by a plasma sample under test conditions, represents the balance between procoagulant and anticoagulant factors. Although this test comes the closest to in vivo conditions, it still requires more research and validation (72).
DISCUSSION
Contrary to the paradigm in which cirrhosis is considered to be disease in which patients tend primarily towards bleeding, it has now been proven that a state of hypercoagulability can coexist with cirrhosis. This state manifests in microvascular and macrovascular thrombosis which is rarely attributed to this condition. Parallel to these findings it has been postulated that the coagulation system in cirrhotic patients is in a state of "rebalance." This is poorer balance than that found in healthy people, but it is capable of adequate functioning even in situations of significant stress for as long as normal platelet hematocrit counts remain normal for the patient's condition. Since the tests that are normally used to evaluate coagulation balances are centered on the procoagulant abnormalities, they do not show this "rebalance" and should be not considered useful tools for the prediction of bleeding or other thrombotic complications. For this reason they are not appropriate decision making tools for cirrhosis patients.
REFERENCES
1. asili S, Raparelli V, Violi F. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? Yes. Eur J Intern Med 2010; 21(2): 62-4.
2. Tripodi A, Primignani M, Mannucci PM. Abnormalities of hemostasis and bleeding in chronic liver disease: the paradigm is challenged. Intern Emerg Med 2010; 5(1): 7-12.
3. Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost 2004; 91(2): 267-75.
4. Northup PG. Hypercoagulation in liver disease. Clin Liver Dis 2009; 13(1): 109-16.
5. Tripodi A. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? No. Eur J Intern Med 2010; 21(2): 65-9.
6. Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost 2006; 32(Suppl 1): 32-8.
7. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol 2007; 27(8): 1687-93.
8. Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci 2006; 11: 1349-59.
9. Monroe DM, Hoffman M, Roberts HR. Transmission of a procoagulant signal from tissue factor-bearing cell to platelets. Blood Coagul Fibrinolysis 1996; 7(4): 459-64.
10. Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol 1999; 19(1):170-7.
11. Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost 2003; 1(7): 1504-14.
12. Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006; 26(1): 41-8.
13. Crawley JT, Zanardelli S, Chion CK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemost 2007; 5(Suppl 1): 95-101.
14. Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost 2001; 38(4 Suppl 12): 958-65.
15. Hoffman MM, Monroe DM. Rethinking the coagulation cascade. Curr Hematol Rep 2005; 4(5): 391-6.
16. Esmon CT. The protein C pathway. Chest 2003; 124(3 Suppl): 26S-32S.
17. Dahlbäck B, Stenflo J. Regulatory mechanisms in hemostasis: natural anticoagulants. En: Hoffman R, Furie B, Benz Jr. EJ, McGlave P, Silberstein LE, Shattil SJ, editors. Hematology: basic principles and practice. 5 edition. Philadelphia: Churchill, Livingstone: Elsevier; 2009. p. 1843-9.
18. Senzolo M, Burra P, Cholongitas E, Burroughs AK. New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol 2006; 12(48): 7725-36.
19. Lisman T, Leebeek FW. Hemostatic alterations in liver disease: a review on pathophysiology, clinical consequences, and treatment. Dig Surg 2007 24(4): 250-8.
20. Goulis J, Chau TN, Jordan S, Mehta AB, Watkinson A, Rolles K, et al. Thrombopoietin concentrations are low in patients with cirrhosis and thrombocytopenia and are restored after orthotopic liver transplantation. Gut 1999; 44(5): 754-8.
21. Kajihara M, Kato S, Okazaki Y, Kawakami Y, Ishii H, Ikeda Y, et al. A role of autoantibody-mediated platelet destruction in thrombocytopenia in patients with cirrhosis. Hepatology 2003; 37(6): 1267-76.
22. Nagamine T, Ohtuka T, Takehara K, Arai T, Takagi H, Mori M. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol 1996; 24(2): 135-40.
23. Feistauer SM, Penner E, Mayr WR, Panzer S. Target platelet antigens of autoantibodies in patients with primary biliary cirrhosis. Hepatology 1997; 25(6): 1343-5.
24. Levine RF, Spivak JL, Meagher RC, Sieber F. Effect of ethanol on thrombopoiesis. Br J Haematol 1986; 62(2): 345-54.
25. Tripodi A. Hemostasis abnormalities in liver cirrhosis: myth or reality? Pol Arch Med Wewn 2008; 118(7-8): 445-8.
26. Escolar G, Cases A, Vinas M, Pino M, Calls J, Cirera I, et al. Evaluation of acquired platelet dysfunctions in uremic and cirrhotic patients using the platelet function analyzer (PFA-100 ): influence of hematocrit elevation. Haematologica 1999; 84(7): 614-9.
27. Pasche B, Ouimet H, Francis S, Loscalzo J. Structural changes in platelet glycoprotein IIb/IIIa by plasmin: determinants and functional consequences. Blood 1994; 83(2): 404-14.
28. Cahill PA, Redmond EM, Sitzmann JV. Endothelial dysfunction in cirrhosis and portal hypertension. Pharmacol Ther 2001; 89(3): 273-93.
29. Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006; 44(1): 53-61.
30. Kerr R. New insights into haemostasis in liver failure. Blood Coagul Fibrinolysis 2003; 14(Suppl 1): S43-5.
31. Pluta A, Gutkowski K, Hartleb M. Coagulopathy in liver diseases. Adv Med Sci 2010; 55(1): 16-21.
32. Blanchard RA, Furie BC, Jorgensen M, Kruger SF, Furie B. Acquired vitamin K-dependent carboxylation deficiency in liver disease. N Engl J Med 1981; 305(5): 242-8.
33. Francis JL, ArmB DJ. Fibrinogen-bound sialic acid levels in the dysfibrinogenaemia of liver disease. Haemostasis 1982; 11(4): 215-22.
34. Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol 2007; 46(4): 727-33.
35. Leiper K, Croll A, Booth NA, Moore NR, Sinclair T, Bennett B. Tissue plasminogen activator, plasminogen activator inhibitors, and activator-inhibitor complex in liver disease. J Clin Pathol 1994; 47(3): 214-7.
36. Lisman T, Leebeek FW, Mosnier LO, Bouma BN, Meijers JC, Janssen HL, et al. Thrombin-activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology 2001; 121(1): 131-9.
37. Colucci M, Binetti BM, Branca MG, Clerici C, Morelli A, Semeraro N, et al. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology 2003; 38(1): 230-7.
38. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol 2002; 37(2): 280-7.
39. Vukovich T, Teufelsbauer H, Fritzer M, Kreuzer S, Knoflach P. Hemostasis activation in patients with liver cirrhosis. Thromb Res 1995; 77(3): 271-8.
40. Artiko V, Obradovic V, Petrovic M, Perisic M, Stojkovic M, Sobic-Saranovic D, et al. Hepatic radionuclide angiography and Doppler ultrasonography in the detection and assessment of vascular disturbances in the portal system. Hepatogastroenterology 2007; 54(75): 892-7.
41. Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl 2005; 11(9): 1107-11.
42. Fallon MB. Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary syndrome. J Clin Gastroenterol 2005; 39(Suppl 2): S138-42.
43. Anstee QM, Wright M, Goldin R, Thursz MR. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis 2009; 13(1): 117-26.
44. Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol 2010; 53(2): 362-71.
45. Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, Reddy KR, et al. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology 2006; 44(4): 1039-46.
46. Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood 2001; 98(9): 2730-5.
47. Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005; 41(3): 553-8.
48. Tripodi A, Primignani M, Chantarangkul V, Dell'Era A, Clerici M, de Franchis R, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology 2009; 137(6): 2105-11.
49. Ewe K. Bleeding after liver biopsy does not correlate with indices of peripheral coagulation. Dig Dis Sci 1981; 26(5): 388-93.
50. Segal JB, Dzik WH. Paucity of studies to support that abnormal coagulation test results predict bleeding in the setting of invasive procedures: an evidence-based review. Transfusion 2005; 45(9): 1413-25.
51. Boks AL, Brommer EJ, Schalm SW, Van Vliet HH. Hemostasis and fibrinolysis in severe liver failure and their relation to hemorrhage. Hepatology 1986; 6(1): 79-86.
52. Tellez-Avila FI, Chavez-Tapia NC, Torre-Delgadillo A. [Coagulation disorders in cirrhosis]. Rev Invest Clin 2007; 59(2): 153-60.
53. Pivalizza EG, Warters RD, Gebhard R. Desmopressin before liver transplantation. Can J Anaesth 2003; 50(7): 748-9.
54. Wong AY, Irwin MG, Hui TW, Fung SK, Fan ST, Ma ES. Desmopressin does not decrease blood loss and transfusion requirements in patients undergoing hepatectomy. Can J Anaesth 2003; 50(1): 14-20.
55. Dahlbäck B. Progress in the understanding of the protein C anticoagulant pathway. Int J Hematol 2004; 79(2): 109-16.
56. Tripodi A, Primignani M, Chantarangkul V, Clerici M, Dell'Era A, Fabris F, et al. Thrombin generation in patients with cirrhosis: the role of platelets. Hepatology 2006; 44(2): 440-5.
57. Holland LL, Brooks JP. Toward rational fresh frozen plasma transfusion: The effect of plasma transfusion on coagulation test results. Am J Clin Pathol 2006; 26(1): 133-9.
58. Lisman T, Adelmeijer J, de Groot PG, Janssen HL, Leebeek FW. No evidence for an intrinsic platelet defect in patients with liver cirrhosis--studies under flow conditions. J Thromb Haemost 2006; 4(9): 2070-2.
59. Kirkwood TB. Calibration of reference thromboplastins and standardisation of the prothrombin time ratio. Thromb Haemost 1983; 49(3): 238-44.
60. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33(2): 464-70.
61. Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KV, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2): 353-8.
62. Arjal R, Trotter JF. International normalized ratio of prothrombin time in the model for end-stage liver disease score: an unreliable measure. Clin Liver Dis 2009; 13(1): 67-71.
63. Tripodi A, Chantarangkul V, Primignani M, Fabris F, Dell'Era A, Sei C, et al. The international normalized ratio calibrated for cirrhosis (INR(liver)) normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology 2007; 46(2): 520-7.
64. Kovacs MJ, Wong A, MacKinnon K, Weir K, Keeney M, Boyle E, et al. Assessment of the validity of the INR system for patients with liver impairment. Thromb Haemost 1994; 71(6): 727-30.
65. Denson KW, Reed SV, Haddon ME, Woodhams B, Brucato C, Ruiz J. Comparative studies of rabbit and human recombinant tissue factor reagents. Thromb Res 1999; 94(4): 255-61.
66. Robert A, Chazouillères O. Prothrombin time in liver failure: time, ratio, activity percentage, or international normalized ratio? Hepatology 1996; 24(6): 1392-4.
67. Kamath PS, Kim WR. The international normalized ratio of prothrombin time in the model for end-stage liver disease score: a reliable measure. Clin Liver Dis 2009; 13(1): 63-6.
68. Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apher Sci 2003; 28(3): 307-17.
69. Kundu SK, Heilmann EJ, Sio R, Garcia C, Davidson RM, Ostgaard RA. Description of an in vitro platelet function analyzer--PFA-100. Semin Thromb Hemost 1995; 21(Suppl 2): 106-12.
70. Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 2003; 33(1): 4-15.
71. Chantarangkul V, Clerici M, Bressi C, Giesen PL, Tripodi A. Thrombin generation assessed as endogenous thrombin potential in patients with hyper- or hypo-coagulability. Haematologica 2003; 88(5): 547-54.
72. Tripodi A. Tests of coagulation in liver disease. Clin Liver Dis 2009; 13(1): 55-61.
1. Basili S, Raparelli V, Violi F. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? Yes. Eur J Intern Med 2010; 21(2): 62-4. [ Links ]
2. Tripodi A, Primignani M, Mannucci PM. Abnormalities of hemostasis and bleeding in chronic liver disease: the paradigm is challenged. Intern Emerg Med 2010; 5(1): 7-12. [ Links ]
3. Hollestelle MJ, Geertzen HG, Straatsburg IH, van Gulik TM, van Mourik JA. Factor VIII expression in liver disease. Thromb Haemost 2004; 91(2): 267-75. [ Links ]
4. Northup PG. Hypercoagulation in liver disease. Clin Liver Dis 2009; 13(1): 109-16. [ Links ]
5. Tripodi A. The coagulopathy of chronic liver disease: is there a causal relationship with bleeding? No. Eur J Intern Med 2010; 21(2): 65-9. [ Links ]
6. Roberts HR, Hoffman M, Monroe DM. A cell-based model of thrombin generation. Semin Thromb Hemost 2006; 32(Suppl 1): 32-8. [ Links ]
7. Mackman N, Tilley RE, Key NS. Role of the extrinsic pathway of blood coagulation in hemostasis and thrombosis. Arterioscler Thromb Vasc Biol 2007; 27(8): 1687-93. [ Links ]
8. Gomez K, McVey JH. Tissue factor initiated blood coagulation. Front Biosci 2006; 11: 1349-59. [ Links ]
9. Monroe DM, Hoffman M, Roberts HR. Transmission of a procoagulant signal from tissue factor-bearing cell to platelets. Blood Coagul Fibrinolysis 1996; 7(4): 459-64. [ Links ]
10. Oliver JA, Monroe DM, Roberts HR, Hoffman M. Thrombin activates factor XI on activated platelets in the absence of factor XII. Arterioscler Thromb Vasc Biol 1999; 19(1):170-7. [ Links ]
11. Mann KG, Brummel K, Butenas S. What is all that thrombin for? J Thromb Haemost 2003; 1(7): 1504-14. [ Links ]
12. Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006; 26(1): 41-8. [ Links ]
13. Crawley JT, Zanardelli S, Chion CK, Lane DA. The central role of thrombin in hemostasis. J Thromb Haemost 2007; 5(Suppl 1): 95-101. [ Links ]
14. Hoffman M, Monroe DM 3rd. A cell-based model of hemostasis. Thromb Haemost 2001; 38(4 Suppl 12): 958-65. [ Links ]
15. Hoffman MM, Monroe DM. Rethinking the coagulation cascade. Curr Hematol Rep 2005; 4(5): 391-6. [ Links ]
16. Esmon CT. The protein C pathway. Chest 2003; 124(3 Suppl): 26S-32S. [ Links ]
17. Dahlbäck B, Stenflo J. Regulatory mechanisms in hemostasis: natural anticoagulants. En: Hoffman R, Furie B, Benz Jr. EJ, McGlave P, Silberstein LE, Shattil SJ, editors. Hematology: basic principles and practice. 5 edition. Philadelphia: Churchill, Livingstone: Elsevier; 2009. p. 1843-9. [ Links ]
18. New insights into the coagulopathy of liver disease and liver transplantation. World J Gastroenterol 2006; 12(48): 7725-36. [ Links ]
19. Lisman T, Leebeek FW. Hemostatic alterations in liver disease: a review on pathophysiology, clinical consequences, and treatment. Dig Surg 2007 24(4): 250-8. [ Links ]
20. Goulis J, Chau TN, Jordan S, Mehta AB, Watkinson A, Rolles K, et al. Thrombopoietin concentrations are low in patients with cirrhosis and thrombocytopenia and are restored after orthotopic liver transplantation. Gut 1999; 44(5): 754-8. [ Links ]
21. Kajihara M, Kato S, Okazaki Y, Kawakami Y, Ishii H, Ikeda Y, et al. A role of autoantibody-mediated platelet destruction in thrombocytopenia in patients with cirrhosis. Hepatology 2003; 37(6): 1267-76. [ Links ]
22. Nagamine T, Ohtuka T, Takehara K, Arai T, Takagi H, Mori M. Thrombocytopenia associated with hepatitis C viral infection. J Hepatol 1996; 24(2): 135-40. [ Links ]
23. Feistauer SM, Penner E, Mayr WR, Panzer S. Target platelet antigens of autoantibodies in patients with primary biliary cirrhosis. Hepatology 1997; 25(6): 1343-5. [ Links ]
24. Levine RF, Spivak JL, Meagher RC, Sieber F. Effect of ethanol on thrombopoiesis. Br J Haematol 1986; 62(2): 345-54. [ Links ]
25. Tripodi A. Hemostasis abnormalities in liver cirrhosis: myth or reality? Pol Arch Med Wewn 2008; 118(7-8): 445-8. [ Links ]
26. Escolar G, Cases A, Vinas M, Pino M, Calls J, Cirera I, et al. Evaluation of acquired platelet dysfunctions in uremic and cirrhotic patients using the platelet function analyzer (PFA-100 ): influence of hematocrit elevation. Haematologica 1999; 84(7): 614-9. [ Links ]
27. Pasche B, Ouimet H, Francis S, Loscalzo J. Structural changes in platelet glycoprotein IIb/IIIa by plasmin: determinants and functional consequences. Blood 1994; 83(2): 404-14. [ Links ]
28. Cahill PA, Redmond EM, Sitzmann JV. Endothelial dysfunction in cirrhosis and portal hypertension. Pharmacol Ther 2001; 89(3): 273-93. [ Links ]
29. Lisman T, Bongers TN, Adelmeijer J, Janssen HL, de Maat MP, de Groot PG, et al. Elevated levels of von Willebrand Factor in cirrhosis support platelet adhesion despite reduced functional capacity. Hepatology 2006; 44(1): 53-61. [ Links ]
30. Kerr R. New insights into haemostasis in liver failure. Blood Coagul Fibrinolysis 2003; 14(Suppl 1): S43-5. [ Links ]
31. Pluta A, Gutkowski K, Hartleb M. Coagulopathy in liver diseases. Adv Med Sci 2010; 55(1): 16-21. [ Links ]
32. Blanchard RA, Furie BC, Jorgensen M, Kruger SF, Furie B. Acquired vitamin K-dependent carboxylation deficiency in liver disease. N Engl J Med 1981; 305(5): 242-8. [ Links ]
33. Francis JL, Armstrong DJ. Fibrinogen-bound sialic acid levels in the dysfibrinogenaemia of liver disease. Haemostasis 1982; 11(4): 215-22. [ Links ]
34. Tripodi A, Mannucci PM. Abnormalities of hemostasis in chronic liver disease: reappraisal of their clinical significance and need for clinical and laboratory research. J Hepatol 2007; 46(4): 727-33. [ Links ]
35. Leiper K, Croll A, Booth NA, Moore NR, Sinclair T, Bennett B. Tissue plasminogen activator, plasminogen activator inhibitors, and activator-inhibitor complex in liver disease. J Clin Pathol 1994; 47(3): 214-7. [ Links ]
36. Lisman T, Leebeek FW, Mosnier LO, Bouma BN, Meijers JC, Janssen HL, et al. Thrombin-activatable fibrinolysis inhibitor deficiency in cirrhosis is not associated with increased plasma fibrinolysis. Gastroenterology 2001; 121(1): 131-9. [ Links ]
37. Colucci M, Binetti BM, Branca MG, Clerici C, Morelli A, Semeraro N, et al. Deficiency of thrombin activatable fibrinolysis inhibitor in cirrhosis is associated with increased plasma fibrinolysis. Hepatology 2003; 38(1): 230-7. [ Links ]
38. Lisman T, Leebeek FW, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol 2002; 37(2): 280-7. [ Links ]
39. Vukovich T, Teufelsbauer H, Fritzer M, Kreuzer S, Knoflach P. Hemostasis activation in patients with liver cirrhosis. Thromb Res 1995; 77(3): 271-8. [ Links ]
40. Artiko V, Obradovic V, Petrovic M, Perisic M, Stojkovic M, Sobic-Saranovic D, et al. Hepatic radionuclide angiography and Doppler ultrasonography in the detection and assessment of vascular disturbances in the portal system. Hepatogastroenterology 2007; 54(75): 892-7. [ Links ]
41. Kawut SM, Taichman DB, Ahya VN, Kaplan S, Archer-Chicko CL, Kimmel SE, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transpl 2005; 11(9): 1107-11. [ Links ]
42. Fallon MB. Mechanisms of pulmonary vascular complications of liver disease: hepatopulmonary syndrome. J Clin Gastroenterol 2005; 39(Suppl 2): S138-42. [ Links ]
43. Anstee QM, Wright M, Goldin R, Thursz MR. Parenchymal extinction: coagulation and hepatic fibrogenesis. Clin Liver Dis 2009; 13(1): 117-26. [ Links ]
44. Lisman T, Caldwell SH, Burroughs AK, Northup PG, Senzolo M, Stravitz RT, et al. Hemostasis and thrombosis in patients with liver disease: the ups and downs. J Hepatol 2010; 53(2): 362-71. [ Links ]
45. Caldwell SH, Hoffman M, Lisman T, Macik BG, Northup PG, Reddy KR, et al. Coagulation disorders and hemostasis in liver disease: pathophysiology and critical assessment of current management. Hepatology 2006; 44(4): 1039-46. [ Links ]
46. Mannucci PM, Canciani MT, Forza I, Lussana F, Lattuada A, Rossi E. Changes in health and disease of the metalloprotease that cleaves von Willebrand factor. Blood 2001; 98(9): 2730-5. [ Links ]
47. Tripodi A, Salerno F, Chantarangkul V, Clerici M, Cazzaniga M, Primignani M, et al. Evidence of normal thrombin generation in cirrhosis despite abnormal conventional coagulation tests. Hepatology 2005; 41(3): 553-8. [ Links ]
48. Tripodi A, Primignani M, Chantarangkul V, Dell'Era A, Clerici M, de Franchis R, et al. An imbalance of pro- vs anti-coagulation factors in plasma from patients with cirrhosis. Gastroenterology 2009; 137(6): 2105-11. [ Links ]
49. Ewe K. Bleeding after liver biopsy does not correlate with indices of peripheral coagulation. Dig Dis Sci 1981; 26(5): 388-93. [ Links ]
50. Segal JB, Dzik WH. Paucity of studies to support that abnormal coagulation test results predict bleeding in the setting of invasive procedures: an evidence-based review. Transfusion 2005; 45(9): 1413-25. [ Links ]
51. Boks AL, Brommer EJ, Schalm SW, Van Vliet HH. Hemostasis and fibrinolysis in severe liver failure and their relation to hemorrhage. Hepatology 1986; 6(1): 79-86. [ Links ]
52. Tellez-Avila FI, Chavez-Tapia NC, Torre-Delgadillo A. [Coagulation disorders in cirrhosis]. Rev Invest Clin 2007; 59(2): 153-60. [ Links ]
53. Pivalizza EG, Warters RD, Gebhard R. Desmopressin before liver transplantation. Can J Anaesth 2003; 50(7): 748-9. [ Links ]
54. Wong AY, Irwin MG, Hui TW, Fung SK, Fan ST, Ma ES. Desmopressin does not decrease blood loss and transfusion requirements in patients undergoing hepatectomy. Can J Anaesth 2003; 50(1): 14-20. [ Links ]
55. Dahlbäck B. Progress in the understanding of the protein C anticoagulant pathway. Int J Hematol 2004; 79(2): 109-16. [ Links ]
56. Tripodi A, Primignani M, Chantarangkul V, Clerici M, Dell'Era A, Fabris F, et al. Thrombin generation in patients with cirrhosis: the role of platelets. Hepatology 2006; 44(2): 440-5. [ Links ]
57. Holland LL, Brooks JP. Toward rational fresh frozen plasma transfusion: The effect of plasma transfusion on coagulation test results. Am J Clin Pathol 2006; 26(1): 133-9. [ Links ]
58. Lisman T, Adelmeijer J, de Groot PG, Janssen HL, Leebeek FW. No evidence for an intrinsic platelet defect in patients with liver cirrhosis--studies under flow conditions. J Thromb Haemost 2006; 4(9): 2070-2. [ Links ]
59. Kirkwood TB. Calibration of reference thromboplastins and standardisation of the prothrombin time ratio. Thromb Haemost 1983; 49(3): 238-44. [ Links ]
60. Kamath PS, Wiesner RH, Malinchoc M, Kremers W, Therneau TM, Kosberg CL, et al. A model to predict survival in patients with end-stage liver disease. Hepatology 2001; 33(2): 464-70. [ Links ]
61. Dunn W, Jamil LH, Brown LS, Wiesner RH, Kim WR, Menon KV, et al. MELD accurately predicts mortality in patients with alcoholic hepatitis. Hepatology 2005; 41(2): 353-8. [ Links ]
62. Arjal R, Trotter JF. International normalized ratio of prothrombin time in the model for end-stage liver disease score: an unreliable measure. Clin Liver Dis 2009; 13(1): 67-71. [ Links ]
63. Tripodi A, Chantarangkul V, Primignani M, Fabris F, Dell'Era A, Sei C, et al. The international normalized ratio calibrated for cirrhosis (INR(liver)) normalizes prothrombin time results for model for end-stage liver disease calculation. Hepatology 2007; 46(2): 520-7. [ Links ]
64. Kovacs MJ, Wong A, MacKinnon K, Weir K, Keeney M, Boyle E, et al. Assessment of the validity of the INR system for patients with liver impairment. Thromb Haemost 1994; 71(6): 727-30. [ Links ]
65. Denson KW, Reed SV, Haddon ME, Woodhams B, Brucato C, Ruiz J. Comparative studies of rabbit and human recombinant tissue factor reagents. Thromb Res 1999; 94(4): 255-61. [ Links ]
66. Robert A, Chazouillères O. Prothrombin time in liver failure: time, ratio, activity percentage, or international normalized ratio? Hepatology 1996; 24(6): 1392-4. [ Links ]
67. Kamath PS, Kim WR. The international normalized ratio of prothrombin time in the model for end-stage liver disease score: a reliable measure. Clin Liver Dis 2009; 13(1): 63-6. [ Links ]
68. Rand ML, Leung R, Packham MA. Platelet function assays. Transfus Apher Sci 2003; 28(3): 307-17. [ Links ]
69. Kundu SK, Heilmann EJ, Sio R, Garcia C, Davidson RM, Ostgaard RA. Description of an in vitro platelet function analyzer--PFA-100. Semin Thromb Hemost 1995; 21(Suppl 2): 106-12. [ Links ]
70. Hemker HC, Giesen P, Al Dieri R, Regnault V, de Smedt E, Wagenvoord R, et al. Calibrated automated thrombin generation measurement in clotting plasma. Pathophysiol Haemost Thromb 2003; 33(1): 4-15. [ Links ]
71. Chantarangkul V, Clerici M, Bressi C, Giesen PL, Tripodi A. Thrombin generation assessed as endogenous thrombin potential in patients with hyper- or hypo-coagulability. Haematologica 2003; 88(5): 547-54. [ Links ]
72. Tripodi A. Tests of coagulation in liver disease. Clin Liver Dis 2009; 13(1): 55-61. [ Links ]

