










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.28 no.2 Bogotá Apr./June 2013
Linfangiectasia intestinal: Reporte de un caso
Wilson Daza Carreño, MD (1), Luz Marina Mejía Cardona, MD (2), Lina Eugenia Jaramillo Barberi, MD (3), María Carolina Uribe G., MD (4)
(1) Gastroenterólogo pediatra, Magíster Nutrición Clínica - Director de Gastronutriped y del Posgrado Gastroenterología Pediátrica Universidad El Bosque. Bogotá, Colombia.
(2) Pediatra Intensivista - Unidad de Cuidados Intensivos Instituto de Ortopedia Infantil Roosevelt. Bogotá, Colombia.
(3) Patóloga pediatra - Jefe del Departamento de Patología del Hospital La Misericordia. Bogotá, Colombia.
(4) Fellow de Gastroenterología Pediátrica - Universidad El Bosque. Bogotá, Colombia.
Fecha recibido: 06-08-12 Fecha aceptado: 16-04-13
Resumen
A propósito del caso de un paciente de 7 meses de vida remitido desde Yopal a la ciudad de Bogotá, se revisa el tema de la linfagiectasia intestinal. Esta es una rara enfermedad que involucra los vasos linfáticos intestinales, y origina hipoproteinemia, edemas, ascitis y enteropatía perdedora de proteínas.
Palabras clave
Linfangiectasia intestinal, hipoproteinemia, enteropatía perdedora de proteínas, ascitis, estado hipooncótico.
Caso clínico presentado en el III congreso internacional de Gastroenterología, Hepatología y Nutrición Pediátrica. Realizado en Bogotá, los días 31 de mayo, 1 y 2 de junio de 2012.
INTRODUCCIÓN
La linfangiectasia intestinal es una rara enfermedad que involucra los vasos linfáticos intestinales, con obstrucción del drenaje linfático intestinal, relacionado con defectos congénitos en la formación de los ductos linfáticos; puede ser congénito, secundario o idiopático (20). Las formas congénitas están asociadas, frecuentemente, con anormalidades de los linfáticos a cualquier nivel del organismo, en patologías como Turner, Noonan y Síndrome de Klipper-Trenaunay-Weber. Las formas secundarias están asociadas con pericarditis constrictiva, falla cardíaca, fibrosis retroperitoneal, tuberculosis abdominal, malignidad retroperitoneal, entre otras (8). La linfa, que es rica en proteínas, lípidos y linfocitos, llega al lumen intestinal por ende, cuando existe la anomalía, resulta una enteropatía perdedora de proteínas, esteatorrea y atrofia de vellosidades no específica, con o sin infiltración mononuclear de la lámina propia, involucrando algunas veces el epitelio y sin signos histopatológicos específicos (8, 19, 20). Su pronóstico es infortunado (2). Se desconoce la causa de la enfermedad y su naturaleza es escasamente entendida. Se cree que muchas lesiones son secundarias a procesos asociados con el envejecimiento, anormalidades vasculares e hipoxia tisular. Con frecuencia, se observa un patrón intestinal de linfangiectasias sin que ello implique hallazgos anormales.
CASO CLÍNICO
Se trata de un lactante menor de 7 meses, género masculino, procedente de Yopal (Casanare, Colombia), de descendencia indígena, producto del cuarto embarazo a término sin complicaciones, con sospecha de hepatomegalia por ecografía prenatal. Nace por parto vaginal con un peso de 3.500 gramos; recibió lactancia materna exclusiva por 3 meses, inicia ablactación a los 3 meses de edad con agua de hierbas (al parecer para manejo de "diarrea"); a los 7 meses inicia otros alimentos (caldos, sopas de arroz, huevo y frutas como mandarina y naranja). Al momento de la consulta no había recibido leche de vaca, gluten, pescado ni soya. El esquema de vacunación estaba incompleto para la edad. Consultó por cuadro clínico de 17 días de evolución, consistente con deposiciones líquidas en número de 3 al día, sin moco, ni sangre, acompañadas de distensión abdominal y fiebre no cuantificada. En remisión del Hospital de Casanare describen paraclínicos que muestran anemia y trombocitosis, hiponatremia e hipoalbuminemia; ecografía abdominal con ascitis moderada e infiltración de grasa hepática. Remiten a Bogotá con diagnóstico de desnutrición secundaria. Al ingreso, se encuentra en regular estado general, hidratado, pálido, peso de 7.200 gramos (-1,56 DE según curvas OMS), longitud de 67 cm (-0,90 DE según curvas OMS), con peso para la talla (P/T) en -1,07 DE; abdomen globoso, doloroso a la palpación y con onda ascítica positiva (figura 1).
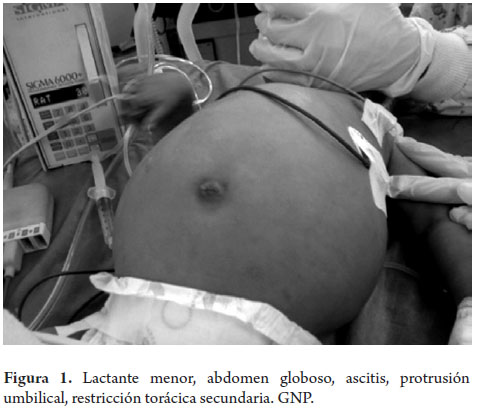
Durante la estancia hospitalaria se documentan proteínas totales y albúmina en descenso, anemia microcítica e hipocrómica, por lo cual inician suplementación con sulfato ferroso, ácido fólico y ácido ascórbico. Al examen físico, se observan lesiones compatibles con micosis en el cuello, por lo que se indicó manejo con clotrimazol. El paciente presenta una evolución tórpida, persistencia de su diarrea y alto gasto (hasta 7 deposiciones al día), hipoalbuminemia (hasta 1,34 mg/dl), aumento progresivo del perímetro abdominal e hipoxemia, requiriendo manejo en Unidad de Cuidado Intensivo pediátrico (UCIP).
Durante su estancia en UCIP, presenta deposiciones pálidas, se documentó hipertensión abdominal, insuficiencia pre renal e hipertensión arterial; deterioro infeccioso debido a neumonía apical derecha e infección de vías urinarias por E. Coli, por lo cual se indicó manejo antibiótico con ampicilina sulbactam y se suspendió el sulfato ferroso, además, se le dio manejo con vitamina K por tiempo de protrombina prolongado. La distensión abdominal fue progresiva, se determinó ascitis moderada e infiltración de grasa hepática por ecografía abdominal. Por lo anterior, se realizó paracentesis a nivel de fosa ilíaca derecha, obteniéndose líquido de aspecto lechoso, con citoquímico quiloso, pH 8,0, leucocitos 17, hematíes 275, glucosa 278,7, proteínas 3,7, LDH 38,3, colesterol 398 mg/dl, TAG 485 mg/dl; sumado a quiloperitoneo, lo que definió la disminución del aporte hídrico, manejo con espironolactona y nutrición parenteral. Se toma baciloscopia seriada, adenosin deaminasa, cultivo de líquido peritoneal, todos resultaron negativos. Se hizo tomografía axial computarizada de abdomen simple y contrastado, observándose engrosamiento en las paredes del íleon terminal hasta 4 mm y del colon en su porción ascendente, proceso inflamatorio intestinal tanto en intestino delgado como grueso, sin lesión retroperitoneal paraneoplásica (figura 2).
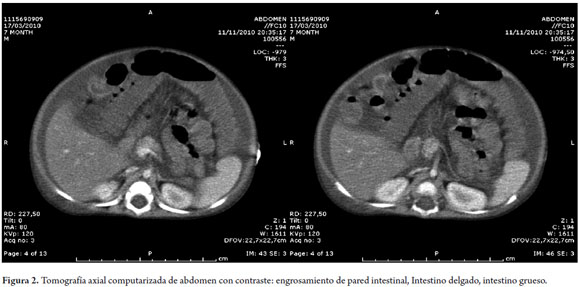
Ante estos hallazgos clínicos y paraclínicos se solicitó valoración por gastroenterólogo pediatra, quien sospecha como una posible linfangiectasia intestinal (LI). Lo anterior, dada la conjunción de compromiso nutricional (desnutrición proteica, Kwashiorkor), diarrea crónica que inició antes de comenzar complementaria, distensión abdominal, hipoalbuminemia, presencia de edemas, ascitis e infecciones asociadas. Se solicitan inmunoglobulinas totales, inmunoglobulinas específicas para toxoplasma, Epstein Bar y citomegalovirus, antígeno de superficie de hepatitis B, VDRL, colesterol total, triglicéridos, ecocardiograma y endoscopia digestiva alta con biopsias para determinar si hay imagen en copos de algodón y dilatación de los vasos linfáticos (LI), así como electrolitos en sudor, con el fin de descartar como diagnósticos diferenciales: fibrosis quística, abetalipoproteinemia, infección o cardiopatía secundaria.
Entre los resultados figuran: inmunoglobulina fracción G, 423 mg/dl (434-1142) que es baja para la edad, inmunoglobulinas E 105 UI/ml (< 10), A 81 mg/dl (15-95), M 54 mg/dl (45-223), iontoforesis en 29 meq/l (negativa para la edad), colesterol total (122,49 mg/dl) y triglicéridos (79 mg/dl) normales, descartando abetalipoproteinemia; inmunoglobulinas específicas para toxoplasmosis IgM 0,3 mg/dl (negativa), citomegalovirus IgM 17,9 mg/dl (positiva), Epstein Bar IgG 19,9 mg/dl (positiva), por lo que se sospecha reactividad cruzada; antígeno de superficie para hepatitis B en 0,19 mg/dl (no reactivo), serología no reactiva y ecocardiograma normal.
Dado el compromiso de filtración glomerular, con hipertensión arterial y diuresis conservada en este paciente con desnutrición crónica edematosa, no se descarta que se esté desarrollando un síndrome hepatorrenal; se considera que requiere manejo agresivo con antibióticos, mantener niveles adecuados de glicemia, manejo hidroelectrolítico y evitar excesiva manipulación por riesgo de infección nosocomial.
Se indicó iniciar nutrición enteral por sonda nasoduodenal con un suplemento nutricional completo específico, que característicamente es bajo en el aporte de triglicéridos de cadena larga, con alto contenido de triglicéridos de cadena media (90%), aporte alto de proteínas, vitaminas y minerales. Al aumentar la nutrición enteral, se fue disminuyendo progresivamente la nutrición parenteral para evitar síndrome de realimentación, brindando proteínas a 1 gr/kg/día con suplementación de vitamina A, 100.000 UI dosis inicial y una segunda dosis a las 2 semanas.
Se realizó endoscopia digestiva alta encontrándose lesiones eritematosas erosivas no sangrantes en estómago, cuerpo y antro con duodeno anormal hasta la tercera porción; por imágenes en copos de algodón que dan aspecto de empedrado de la mucosa en relación con linfangiectasia intestinal primaria (LIP) y gastritis corporoantral eritematosa erosiva, se tomaron biopsias y se enviaron para su análisis con diagnóstico de LIP (figura 3).
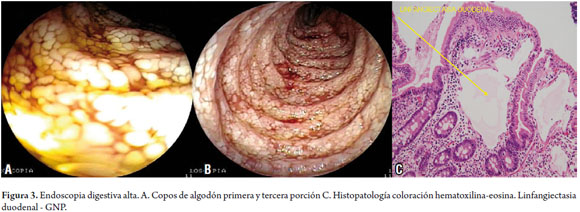
El estudio de patología muestra múltiples cavidades dilatadas en la lámina propia del duodeno, de paredes delgadas delineadas por endotelio plano, y ocupadas en su luz por plasma, que permiten identificarlas como vasos linfáticos. Las vellosidades muestran engrosamiento y aspecto "aporrado" y hay infiltrado linfoplasmocitario con mezcla de eosinófilos. Con estos hallazgos se confirma la sospecha diagnóstica de linfangiectasia intestinal y duodenitis crónica.
Con el manejo instaurado, la evolución clínica resultó acorde a lo esperado, con disminución progresiva de edemas, diarrea y ascitis con mejoría hasta egreso hospitalario y traslado a ciudad de origen.
DISCUSIÓN
La enteropatía perdedora de proteínas es una condición rara, caracterizada por la pérdida a través del tracto gastrointestinal, que lleva a una disminución progresiva de los niveles séricos. Su incidencia y prevalencia son desconocidas (2). La hipoproteinemia conduce a edema, ascitis, efusiones pleurales y cardíacas. Puede ser secundaria a diferentes desórdenes que causan lesión a nivel de la mucosa, como por ejemplo, enfermedad inflamatoria intestinal, neoplasias, falla cardíaca, o anormalidades del sistema linfático como lo es la linfangiectasia intestinal, en nuestro caso (6, 7, 19, 20).
La ascitis quilosa es una entidad bastante rara, aparece a cualquier edad (19); en pediatría lo más común es que se derive de alteraciones congénitas en los linfáticos, y en otros casos, puede ser secundaria a trauma u obstrucción linfática extrínseca (masas abdominales). Lo más habitual es que se manifieste como una enteropatía con pérdida de proteínas de larga evolución, variando en sus manifestaciones y gravedad (19, 20).
En 1961, Waldmann describe la linfangiectasia intestinal (LI) (2, 7, 13); los síntomas pueden variar desde diarrea crónica en los primeros meses de vida, malabsorción, desnutrición, edemas, hipoalbuminemia, linfopenia, hipogamaglobulinemia, infecciones asociadas e incluso provocar la muerte (2). Los grados de severidad dependen de la localización anatómica y de la extensión de la anomalía linfática (2, 7, 13, 19).
La linfagiectasia intestinal (LI) es una enfermedad poco común, se presenta en niños y adultos (19). Consiste en una dilatación de los vasos linfáticos intestinales que origina un trastorno del drenaje linfático. Puede ser primaria (LIP), por defecto congénito en la formación de los canales linfáticos o secundaria a una enfermedad de base, radioterapia, quimioterapia o cirugías (2, 8, 20). Ocasiona un aumento en la presión de los linfáticos que lleva a la pérdida de linfa hacia el lumen intestinal. Esta pérdida, rica en proteínas y lípidos, condiciona enteropatía perdedora de proteínas, asociada a disminución de la circulación de linfocitos desde el intestino hacia la periferia y esteatorrea; la causa de la LIP es desconocida (2, 8, 19, 20).
Además de estas pérdidas, también existe malabsorción de quilomicrones y de vitaminas liposolubles, con hallazgos característicos y evidenciados en la paracentesis tal como aconteció en nuestro paciente, en relación con quiloperitoneo (2).
En la literatura se describe asociación entre hijos y padres consanguíneos, lo cual sugiere origen genético con transmisión autosómica recesiva. El pronóstico de este tipo de diarrea intratable es pobre, la mayoría de los pacientes fallecen entre los 2 y 5 años de edad, algunos de ellos con estadios tempranos de enfermedad hepática; es responsable de aproximadamente 30 a 40% de los casos de sangrado gastrointestinal oscuro (20).
En la linfangiectasia de causa secundaria, la dilatación de los linfáticos es causada por obstrucción de los vasos o por presión linfática elevada secundaria al aumento en la presión venosa central. La obstrucción se puede presentar en pacientes con enfermedad inflamatoria intestinal, sarcoidosis o linfoma, mientras que el aumento de la presión linfática acontece en pacientes con falla cardíaca o pericarditis congestiva. Se ha descrito su asociación con algunos síndromes como: Von Recklinghausen, Turner, Noonan, Klipper Trenaunay y Hennekan (2, 8). Otro grupo importante de pacientes con linfangiectasia secundaria es aquel con antecedente de cirugía de Fontan por cardiopatía congénita compleja, en cuyo caso, es una complicación que amenaza la vida, ocurre en 4 a 13% de estos pacientes, se asocia con cifras altas de mortalidad y supervivencia a 5 años de 46 a 50%; su fisiopatología no está completamente entendida. La presión venosa sistémica elevada, junto a la dilatación de los linfáticos y pérdida de proteínas secundaria, desempeña un papel importante. Ninguno de estos casos se relaciona con la patología de nuestro paciente, no obstante, deben tenerse en cuenta en el planteamiento del diagnóstico diferencial (7, 8, 10).
Existen reportes con relación a la enteropatía perdedora de proteínas asociada a infección por citomegalovirus (CMV) y la enfermedad de Menetrier, caracterizada por hiperplasia gástrica foveolar, usualmente con curso benigno. En nuestro caso, llama la atención la Ig M positiva para citomegalovirus aunque sin hallazgos característicos en la endoscopia ni signos clínicos sugestivos de esta entidad. La mayoría de casos se ha informado en adultos (2, 3, 20).
El grado de afectación intestinal es variable y en ocasiones se deben hacer biopsias múltiples por medio de laparoscopia. El paciente en estudio evidencia un cuadro clínico clásico que se corresponde con diarrea crónica, hipoalbuminemia, ascitis y edemas periféricos secundarios, por formación de tercer espacio (2, 7).
El diagnóstico se establece con base en el conjunto de hallazgos clínicos, radiológicos, endoscópicos e histológicos de la alteración linfática (2, 8); teniendo en cuenta las tres características cardinales, dadas por hipoproteinemia, linfocitopenia y presencia de lesiones anatómicas características. Los estudios radiológicos son poco útiles para diferenciar la causa, sin embargo, en nuestro caso se descarta la presencia de lesiones retroperitoneales o masas que pudieran explicar la causa de la dilatación de los linfáticos secundaria a obstrucción, lo cual se correlaciona con los reportes de la literatura (2, 7, 20).
La medición de alfa 1-antitripsina es una de las herramientas que pueden utilizarse para documentar la pérdida de proteínas a nivel intestinal (13, 14); es una glicoproteína sintetizada por el hígado, que corresponde a 80% de la fracción alfa 1- globulina sérica y alrededor de 4% del contenido proteico del suero; no es degradada por las proteasas intestinales, ni es secretada ni absorbida, por lo que su determinación sirve como medida de la exudación de albúmina hacia la luz intestinal, constituyéndose en un método fácil y no invasivo (20). La detección precoz por medio de la investigación de α1 - Atfecal o AC α1 - AT se ha descrito recientemente en prematuros con linfangiectasia intestinal, herramienta útil en neonatología ante un recién nacido con hipoalbuminemia y edemas periféricos (2, 14, 20). Su medición es un indicador sensible y fiable de pérdida proteica intestinal aunque se discute su especificidad (13, 14, 20). En nuestro paciente, lamentablemente, no se hizo la determinación de alfa 1-antitripsina, para corroborar las evidencias de la literatura mundial.
Las características macroscópicas observadas en la endoscopia digestiva alta consisten en pliegues prominentes (pliegues de Kerchring) con la apariencia blanquecina de las vellosidades, puntos blancos pequeños que no se eliminan con el lavado endoscópico, imagen en "copos de algodón", algunas veces similares a las producidas por acúmulos de grasa y en ocasiones, podría observarse material quiloso en la mucosa (8). La expresión de linfáticos dilatados se confirma por el examen histológico, no obstante, el resultado normal no descarta el diagnóstico por ser una lesión con falta de uniformidad (1, 4, 20).
Si la lesión está limitada a un segmento intestinal, la resección del intestino puede solucionar el problema. Sin embargo, cuando la afectación es difusa, su manejo es complicado ya que persisten pérdidas de proteínas, grasas y linfocitos a través de los linfáticos dilatados. Estas pérdidas generan un estado de malnutrición crónica con inmunodeficiencia asociada (secundaria) que aumenta el riesgo de infecciones, tal como acontece con el paciente-estudio. Siendo la hipogamaglobulinemia y linfopenia secundarias a la pérdida de la linfa (2, 9), la inmunidad celular puede afectarse por la linfopenia grave e incluso, mantenerse en algunos casos; se considera que es secundaria a la mayor pérdida relativa de linfocitos T de vida media larga, en comparación con linfocitos B de vida media más corta y menos susceptibles a la pérdida gastrointestinal de quilo, porque permanecen mayor tiempo en la circulación periférica. Las manifestaciones cutáneas son comunes, esto explica la evolución tórpida de nuestro paciente desde el punto de vista infeccioso y se correlaciona con los reportes en la literatura (9, 10, 13).
El tratamiento se basa en la restricción de ácidos grasos de cadena larga y administración de triglicéridos de cadena media (11), que son de fácil acceso a la circulación enterohepática y obvian su paso a través del sistema linfático alterado (8, 19). Instaurado el tratamiento, se observa mejoría clara en el número y características de las deposiciones, aunque algunos pacientes pueden presentar episodios diarreicos repetidos, que no es el caso del paciente actual. Existe aún cierta incertidumbre respecto de la evolución a largo plazo (2); en algunos pacientes se ha referido edema refractario (11, 12). En los casos refractarios, se describe el uso de octreotide o ácido tranexámico (6, 13, 15-17). Este último "aplica" para pacientes con enfermedad documentada y aumento en la actividad fibrinolítica plasmática que no responde a dieta, aspecto que no concierne al paciente en estudio (15). En cuanto al uso de octreotide, se conoce poco respecto del mecanismo de acción de la somatostatina en la producción de quilo y la presión a nivel del sistema linfático, se cree que disminuye la absorción intestinal de grasas y la concentración de triglicéridos en el conducto torácico así como la reducción del flujo linfático (6). Su utilización resulta controvertida por los efectos secundarios conocidos como náuseas, dolor abdominal, diarrea, flatulencia, hiperglicemia por inhibición de la secreción de insulina y alteración en el crecimiento (6). Con relación al manejo quirúrgico, en las lesiones limitadas a un segmento intestinal la resección puede solucionar el problema (6); se plantea el manejo de shunts para pacientes con quilotórax o ascitis quilosa persistente. Cuando la linfangiectasia intestinal es secundaria a postoperatorio después de un Fontan, se plantea el uso de corticoides o heparina, según sea la situación clínica.
CONCLUSIONES
Hasta el momento, no existen publicaciones de esta patología en Colombia y hay escasa literatura mundial. En pediatría, debe sospecharse cuando el niño consulta por cuadro de diarrea crónica en sus primeros meses de vida, desnutrición, edemas, hipoalbuminemia y linfopenia. El diagnóstico precoz y el manejo nutricional oportuno se anteponen a la pérdida de linfocitos e inmunoglobulinas hacia el lumen intestinal que junto con la pérdida de proteínas, grasas y micronutrientes, condicionan las infecciones a repetición y en ocasiones, hasta la muerte.
REFERENCIAS
1. Van der Meer SB, Forget PP, Willebrand D. Intestinal lymphangiectasia without protein loss in a child with abdominal pain. J Pediatr Gastroenterol Nutr 1990; 10: 246-248. [ Links ]
2. Braamskamp M, Dolman K, Tabbers M. Practice protein losing enteropathy in children. Eur J Pediatr 2010; 169:1179-1185. [ Links ]
3. Hoshina T, Kusuhara K, Saito M, Hara T. Cytomegalovirus-associated protein- Losing enteropathy Resulting from lymphangiectasia in a Immunocompetent child. Jpn J Infect Dis 2009; 62(3): 236-8. [ Links ]
4. Hart MH, Vanderhoof JA, Antonson DL. Failure of blind small bowel biopsy in the diagnosis of intestinal lymphangiectasia. J Pediatr Gastroenterol Nutr 1987; 6(5): 803-5. [ Links ]
5. Macdonald J, Porter V, Scott NW, McNamara D. Small bowel lymphangiectasia and angiodysplasia, a positive association; Novel clinical marker or shared pathophysiology? J Clin Gastroenterol Nutr 2010; 44(9): 610-4. [ Links ]
6. Sari S, Baris Z, Dalgic B. Primary intestinal lymphangiectasia in children: Is octreotide an effective and safe option in the treatment? J Pediatr Gastroenterol Nutr 2010; 51(4): 454-7. [ Links ]
7. Uğuralp S, Mutus M, Kutlu O, Cetin S, Baysal T, Mizrak B. Primary intestinal lymphangiectasia: a rare disease in the differential diagnosis of acute abdomen. J Pediatr Gastroenterol Nutr 2001; 33(4): 508-10. [ Links ]
8. Suresh N, Ganesh R, Sankar J, Sathiyasekaran M. Primary intestinal lymphangiectasia. Indian Pediatr 2009; 46(10): 903-6. [ Links ]
9. Dierselhuis MP, Boelens JJ, Versteegh FG, Weemaes C, Wulffraat NM. Recurrent and opportunistic infections in children with primary intestinal lymphangiectasia. J Pediatr Gastroenterol Nutr 2007; 44(3): 382-5. [ Links ]
10. Strober W, Wochner RD, Carbone PP, Waldmann TA. Intestinal lymphangiectasia: a protein-losing enteropathy with hypogammaglobulinemia, lymphocytopenia and impaired homograft rejection. J Clin Invest 1967; 46(10): 1643-56. [ Links ]
11. Tift WL, Lloyd JK. Intestinal lymphangiectasia. Long-term results with MCT diet. Arch Dis Child 1975; 50(4): 269-76. [ Links ]
12. Donzelli F, Norberto L, Marigo A, Barbato A, Tapparello G, Basso G, Zacchello G. Primary intestinal lymphangiectasia. Comparison between endoscopic and radiological findings. Helv Paediat Acta 1980; 35: 169-175. [ Links ]
13. Molina M, Romero A, Antón S, Sarría J, Prieto G, Polanco I. Linfangiectasia intestinal primaria: evolución a largo plazo. An Esp Pediatr 2001; 54(Supl. 3): 33-35. [ Links ]
14. Strygler B, Nicar MJ, Santangelo WC, Porter JL, Fordtran JS. Alpha 1-antitrypsin excretion in stool in normal subjects and in patients with gastrointestinal disorders. Gastroenterology 1990; 99(5): 1380-7. [ Links ]
15. Mine K, Matsubayashi S, Nakai Y, Nakagawa T. Intestinal lymphangiectasia markedly improved with antiplasmin therapy. Gastroenterology 1989; 96(6): 1596-9. [ Links ]
16. Ballinger AB, Farthing MJ. Octreotide in the treatment of intestinal lymphangiectasia. Eur J Gastroenterol Hepatol 1998; 10(8): 699-702. [ Links ]
17. Kuroiwa G, Takayama T, Sato Y, Takahashi Y, Fujita T, Nobuoka A, Kukitsu T, Kato J, Sakamaki S, Niitsu Y. Primary intestinal lymphangiectasia successfully treated with octreotide. J Gastroenterol 2001; 36(2): 129-32. [ Links ]
18. Vardy PA, Lebenthal E, Shwachman H. Intestinal lymphagiectasia: a reappraisal. Pediatrics 1975; 55(6): 842-51. [ Links ]
19. Perez S, Campuzano M, Bousoño C, Ramos P. Ascitis quilosa congénita con linfangiectasia intestinal. Departamento de pediatría. Hospital universitario central de asturias, Oviedo. Bol pediatr 2007; 47: 132-135 [ Links ]
20. Argüelles M, García N, Pavon B. Tratado de gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. 2011. [ Links ]
