










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.28 no.2 Bogotá Apr./June 2013
A case report of Intestinal Lymphangiectasia
Wilson Daza Carreño, MD (1), Luz Marina Mejía Cardona, MD (2), Lina Eugenia Jaramillo Barberi, MD (3), María Carolina Uribe G., MD (4)
(1) Pediatric Gastroenterologist, Master of Clinical Nutrition - Director of Pediatric Gastroenterology and Nutrition and Director of Graduate Pediatric Gastroenterology Program at the Universidad El Bosque in Bogotá, Colombia.
(2) Pediatric Intensivist and Intensive Care Unit Pediatric Orthopedic Surgeon at the Roosevelt Institute in Bogota, Colombia.
(3) Pediatric Pathologist and Head of the Department of Pathology at Hospital La Misericordia in Bogota, Colombia.
(4) Pediatric Gastroenterology Fellow at the Universidad El Bosque in Bogotá, Colombia.
Received: 06-08-12 Accepted: 16-04-13
Abstract
This is the case report of a 7 month old child from Yopal with intestinal lymphangiectasia who was sent to Bogota. We also review the issue of intestinal lymphangiectasia, a rare disease involving intestinal lymphatic vessels which caused hypoproteinemia, edema, ascites and protein-losing enteropathy.
Keywords
Intestinal lymphangiectasia, hypoproteinemia, protein losing enteropathy, ascites, hypo-oncotic state.
This clinical case was presented in the 3rd International Congress of Pediatric Gastroenterology, Hepatology and Nutrition which took place in Bogota, Colombia from May 31 to June 2, 2012.
INTRODUCTION
Intestinal lymphangiectasia is a rare disease which involves the intestinal lymph vessels including obstruction of lymphatic drainage of the intestines. It is related to congenital, secondary or idiopathic defects in the formation of the lymphatic ducts (20). Congenital forms are frequently associated with lymphatic abnormalities in other areas of the organism in pathologies such as Turner's, Noonan's and Klipper-Trenaunay-Weber Syndromes. Secondary forms are associated with constrictive pericarditis, heart failure, retroperitoneal fibrosis, abdominal tuberculosis, retroperitoneal malignancy and other pathologies (8). Lymph is rich in proteins, lipids and lymphocytes. If there is an anomaly when lymph reaches the intestinal lumen, it results in a protein losing enteropathy, steatorrhea and nonspecific villus atrophy. Mononuclear infiltration of the lamina propria, sometimes involving the epithelium, but without specific histopathological signs, may also be present (8, 19, 20). The prognosis for a patient with intestinal lymphangiectasia is often poor (2). The disease's causes are unknown and its nature is not well understood. It is believed that many lesions are secondary to processes associated with aging, vascular abnormalities and tissue hypoxia. The pattern of intestinal lymphangiectasia is frequently observed without other abnormal findings.
CLINICAL CASE
A 7-month old male from Yopal, Casanare in Colombia) was suspected of having hepatomegaly from a prenatal ultrasound. The child was the fourth born of an indigenous woman all of whose pregnancies went to term without complications. Born by vaginal delivery with a birth weight of 3500 grams, the boy was exclusively breastfed for 3 months. Weaning began at 3 months and the child was given water infused with herbs in an apparent effort to treat diarrhea. At 7 months other foods including broths, rice soups, eggs, tangerines, oranges and other fruit were introduced to his diet. When he was first examined he had not yet received cow's milk, gluten, fish or soy. His vaccination scheme was incomplete for his age. He came to the clinic following 17 days of liquid depositions 3 times a day, without mucus or blood, accompanied by abdominal distension and a fever which had not been measured. After admittance to the hospital in Casanare paraclinical tests showed anemia, thrombocytosis, hyponatremia and hypoalbuminemia. An abdominal ultrasound showed moderate ascites and a fat infiltration in the liver. The patient was sent to Bogota with a diagnosis of secondary malnutrition. Upon admittance the patient's general condition was poor. Although he was hydrated, he was pale and weighed 7200 grams (-1.56 according to WHO graphs) and was 67 cm long (-0.90 according to WHO graphs). His weight to height ratio (W/H) was -1.07. He had a protruding abdomen which was painful when touched and also had a positive ascitic wave (Figure 1).
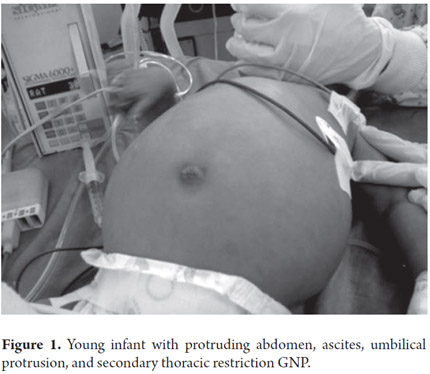
While hospitalized the patient's total protein and albumin levels dropped and hypochromic and microcytic anemia were observed. The patient was given supplements with ferrous sulfate, folic acid and ascorbic acid. Upon physical examination, lesions compatible with neck mycosis were observed and treated with clotrimazole. The patient appeared to be torpid and had persistent diarrhea up to 7 times a day). The patient exhibited hypoalbuminemia (up to 1.34 mg/dl) bloating and hypoxemia which required treatment in the Pediatric Intensive Care Unit (PICU).
In the PICU the child's stools were pale, and he had hypertension and pre-renal insufficiency. He began to suffer infectious deterioration due to right apical pneumonia and an E. coli urinary infection. He was given ampicillin sulbactam and ferrous sulfate was suspended. He was also treated with vitamin k due to prolonged prothrombin. The abdominal distension was progressive. Moderate ascites and hepatic fat infiltration were determined through abdominal ultrasound. For these reasons paracentesis was performed at the right iliac fossa. It obtained a milky liquid with cytochemical chylous. Tests of the liquid showed pH 8.0, leukocytes 17, erythrocytes 275, glucose 278.7, proteins 3.7, LDH 38.3, cholesterol 398 mg/dl, and TAG 485 mg/dl. Chyloperitoneum and dehydration were diagnosed and treated with spironolactone and parenteral nutrition. A serial microscopic examination for acid-fast bacilli (AFB) was negative, as were a test for adenosine deaminase and a peritoneal fluid culture. Simple and contrasted abdominal CAT scans showed a thickening of up to 4mm of the terminal ileum walls and the ascending part of the colon plus an inflammatory process in both the small and large intestine but no paraneoplastic retroperitoneal lesion (see Figure 2).
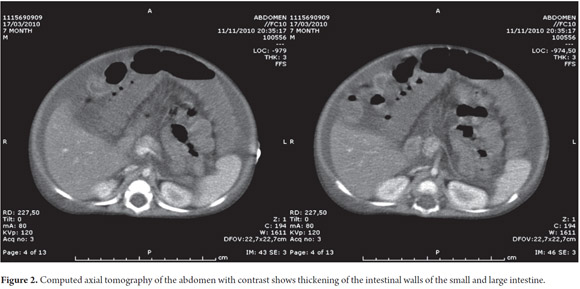
Because of these clinical and paraclinical findings, an assessment from a pediatric gastroenterologist was requested. The doctor suspected possible intestinal lymphangiectasia. This suspicion was based on the combination of nutritional compromise (protein malnutrition, Kwashirkor), chronic diarrhea followed by abdominal distension, hypoalbuminemia, edema, ascites and other associated infections. The patient was tested for total immunoglobulins, immunoglobulins specific for toxoplasma, Epstein bar and cytomegalovirus, surface antigen of Hepatitis B, VDRL, total cholesterol and triglycerides. An echocardiogram and upper digestive endoscopy with biopsies were performed to determine if there was a candy floss image or dilatation of the lymphatic vessels (IL). The child's sweat was tested for electrolytes as part of differential diagnosis to discard other possibilities including cystic fibrosis, abetalipoproteinemia, secondary infection and heart disease.
Results included: immunoglobulin G: 423 mg/dl (434-1142) (low for patient's age), immunoglobulin E: 105 IU/ml (<10), immunoglobulin A 81 mg/dl (15-95), immunoglobulin M 54 mg/dl (45-223), iontophoresis: 29 meq/L (Negative for patient's age), total cholesterol: 122.49 mg/dl, triglycerides: 79mg/dl (Normal, ruling out abetalipoproteinemia), toxoplasmosis specific IgM: 0.3 mg/dl (negative), cytomegalovirus specific IgM: 17.9 mg/dl (positive), Epstein Bar specific IgG: 19.9 mg/dl (positive, which is why cross-reactivity was suspected), hepatitis surface antigen B: 0.19 mg/dl (non-reactive), blood tests were negative and the echocardiogram was normal.
Given the patients compromised glomerular filtration rate, arterial hypertension, oliguria and chronic edematous malnutrition, the possibility of development of hepatorenal syndrome was not discarded. The treatment recommended included aggressive administration of antibiotics, maintenance of adequate glucose levels, management of electrolytes, and avoidance of excessive handling to reduce the risk of nosocomial infection.
Enteral nutrition through a nasal-duodenal probe with a complete specific nutritional supplement was begun. This supplement characteristically has few long chain triglycerides, but is rich in medium chain triglycerides (90%) proteins, vitamins and minerals. As enteral nutrition increased, parenteral nutrition was progressively decreased to avoid refeeding syndrome. The patient was provided with 1gr/kg/day of proteins with supplementary vitamin A at 100,000 UI starting dose with a second dose after two weeks.
Upper digestive endoscopy was performed again. It revealed erosive erythematous lesions in the stomach, corpus and antrum which were not bleeding and showed an abnormal duodenum up to the third portion. Cotton balls images created a cobblestone appearance of the mucosa which is associated with primary intestinal lymphangiectasia and erosive corporal caudal erythematous-antral gastritis. Analysis of biopsies taken confirmed a diagnosis of primary intestinal lymphangiectasia (See Figure 3).
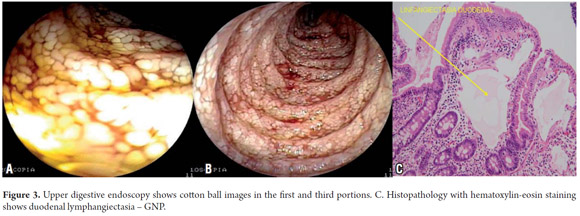
The pathological study revealed numerous dilated cavities in the slide from the duodenum. Its thin walls were lined with flat endothelia filled with plasma which allowed them to be identified as lymphatic vessels. The villi were thickened and club-like with lymphoplasmacytic infiltration mixed with eosinophils. The diagnostic suspicion of intestinal lymphangiectasia and chronic duodenitis was confirmed by these findings.
The treatment established had the expected results of progressively decreasing edema, diarrhea and ascites. The patient improved to the point that he was released from the hospital and returned to his city of origin.
DISCUSSION
Protein losing enteropathies are so rare that their incidence and prevalence are unknown (2). They are characterized by loss of protein through the gastrointestinal tract which leads to progressively decreasing serum levels. Hypoproteinemia leads to edema, ascites, and to pleural and cardiac effusions. A protein losing enteropathy may be secondary to disorders which cause mucosal lesions such as inflammatory bowel disease, neoplasias, heart failure and/or abnormalities of the lymphatic system such as intestinal lymphangiectasia, as in this case (6, 7, 19, 20).
Chylous ascites is another rare entity which can appears at any age (19). The most common causes among pediatric patients are congenital lymphatic alterations, but it may also be secondary to trauma or extrinsic lymphatic obstruction by abdominal masses. Its most common manifestation is through enteropathy with longstanding protein loss which varies in severity as well as in its manifestations (19, 20).
Waldmann described intestinal lymphangiectasia (IL) in 1961 ( 2, 7, 13). Its symptoms may vary from chronic diarrhea in the first months of life to malabsorption, malnutrition, edema, hypoalbuminemia, lymphopenia, hypogammaglobulinemia, associated infections and even death (2). The degree of severity depends on the anatomical location and the extension of the lymphatic anomaly (2, 7, 13, 19).
IL is uncommon in both children and adults (19). It consists of the dilation of intestinal lymphatic vessels which creates a lymphatic drainage syndrome. Although primary intestinal lymphangiectasia (PIL) results from congenital defects in the formation of lymphatic channels, the exact cause of PIL is unknown. Secondary IL is related to a base disease, radiation therapy, chemotherapy or surgery (2, 8, 20). Lymphatic pressure increases leading to loss of lymph into the intestinal lumen. Since lymph is rich in proteins and lipids, this loss leads to longstanding protein loss enteropathy which is associated with decreased circulation of lymphocytes from the intestine to the periphery and steatorrhea (2, 8, 19, 20).
In addition to these losses chylomicrons and liposoluble vitamins are poorly absorbed. This is characteristically evidenced in paracentesis as the case of chyloperitoneum found in our patient (2).
The literature describes an association of IL with consanguineous parents and their children which suggests a genetic origin in recessive autosomal transmission. The prognosis for this type of irritable diarrhea is poor with most patients dying between the ages of two and five years. Some develop early stages of liver disease which is responsible for approximately 30% to 40% cases of occult gastrointestinal bleeding (20).
In secondary IL, lymphatic dilation is caused by the obstruction of vessels or because of elevated lymphatic pressure secondary to increased central venous pressure. Obstructions may develop in patients with inflammatory bowel syndrome, sarcoidosis or lymphoma while increased lymphatic pressure occurs in patients with heart failure or congestive pericarditis. Its has been described in association with Von Recklinghausen's, Turner's, Noonan's, Klipper Trenaunay and Hennekan's syndromes (2, 8). Another important group of patients with secondary lymphangiectasia have undergone Fontan surgery for complex congenital heart disease which is a life-threatening disease. IL occurs in 4% to 13% of these patients among whom it is associated with high mortality rates. Five year survival rates vary from 46% to 50%. Although the pathophysiology of these cases is not completely understood, elevated systemic venous pressure, lymphatic dilation and secondary protein loss play important roles. While none of these cases are related with our patients' pathology, they must be taken into account in the differential diagnosis (7, 8, 10).
Some reports show protein losing enteropathy associated with cytomegalovirus (CMV) infections and Menetrier's disease. These are characterized by foveolar gastric hyperplasia which is usually benign. In our case, the positive test for cytomegalovirus IgM deserves attention, even though there were no characteristic findings from endoscopy or clinical signs pointing to cytomegalovirus. Most of these cases have been reported in adults (2, 3, 20).
Since the intestines can be affected to varying degrees, sometimes many biopsies must be taken laparoscopically. The patient in our study had a classic clinical picture including chronic diarrhea, hypoalbuminemia, ascites and secondary peripheral edema in the third space (2, 7).
The diagnosis is established through clinical, radiological, endoscopic and histological findings regarding the lymphatic alteration (2, 8). The three cardinal characteristics to take into account are hypoproteinemia, lymphocytopenia and the presence of characteristic anatomical lesions. Radiological studies are of little use for discerning the cause of IL, although in our case they allowed us to discard retroperitoneal lesions and masses that might have explained the cause of the lymphatic dilation secondary to an obstruction. This corresponds to experience reported in the literature (2, 7, 20).
Measurement of alpha 1 antitrypsin is one of the tools that may be used to document intestinal protein loss (13, 14). Alpha 1 antitrypsin is a glycoprotein synthesized by the liver which accounts for 80% of the alpha 1- globulin serum fraction and around 4% of serum's protein content. It is not degraded by intestinal proteases, nor can it be secreted or absorbed. For these reasons measurement of alpha 1 antitrypsin serves as an easy and non-invasive method for measuring of the secretion of albumin into the intestine (20). Early detection of alpha 1 antitrypsin through investigation of α1 Atfecal or AC α1 AT has recently been described in premature infants with intestinal lymphangiectasia which should become a useful tool for neonatology for examination of newborns with hypoalbuminemia and peripheral edemas (2, 14, 20). This measurement is a sensitive and reliable indicator of intestinal protein loss, although its specificity is debatable (13, 14, 20). Unfortunately, we did not measure alpha 1- antitrypsin in our study so were unable to corroborate with the world literature in this regard.
Macroscopic characteristics observed in upper digestive endoscopy consist of prominent folds (Kerckring folds) with whitish appearing villi, small white dots that are not eliminated by the endoscopic wash, and "cotton ball" imaging which is sometimes similar to those produced by grease buildup. Occasionally, chylous material can be observed in the mucosa (8). Lymph dilation can be confirmed by histological tests, but a normal result does not rule out a diagnosis of IL since this type of lacks uniformity (1, 4, 20).
If the lesion is limited to one intestinal segment, resection of that segment may solve the problem. However, when the affected area is diffuse, treatment becomes complicated since protein, fat and lymphocyte losses persist throughout the dilated lymphatics. These losses create a state of chronic malnutrition with associated secondary immunodeficiencies that increase the risk of infections as happened to the patient in our study. Since hypogammaglobulinemia and lymphopenia are secondary to the loss of lymph (2, 9), cellular immunity may be affected by severe lymphopenia. In some cases it is considered the second most important factor for loss of T lymphocytes which have longer half-lives than do B lymphocytes. Since T lymphocytes are in peripheral circulation for longer time, they are less susceptible to gastrointestinal loss of chyle than are B lymphocytes. Skin manifestations are common which explains the torpidity of our patient related to infections which correlates to other reports in the literature (9, 10, 13).
Treatment is based on restriction of long chain fatty acids and the administration of medium chain triglycerides (11) which easily access enterohepatic circulation obviating passage through the altered lymphatic system (8, 19). Once treatment has begun, a clear improvement is observed in the number and characteristics of stools, though some patients may suffer repeated episodes of diarrhea (which is not the case with our patient). There is still some uncertainty regarding long-term evolution (2) since refractory edema has been observed in some patients (11, 12). The use of octreotide or tranexamic acid has been described for refractory cases (6, 13, 15, 16, 17). Tranexamic acid is administered to patients with documented disease and increased fibrinolytic activity in the plasma that does not respond to diet. This was not the case in our patient (15). Although little is known about the action mechanism of somatostatins (which octreotide mimics) in the production of chyle and in lymphatic system pressure, it is believed that they decrease intestinal absorption of fats and concentrations of triglycerides in the thoracic duct and also decrease lymphatic flows (6). The use of octreotide is controversial because of secondary effects including nausea, abdominal pain, diarrhea, flatulence, hyperglycemia (resulting from inhibition of insulin secretion) and the alterations in growth (6). Resection may resolve the problem in lesions limited to one intestinal segment (6) while placement of shunts is suggested for patients with persistent chylothorax or chylous ascites. Depending on the clinical situation, the use of corticosteroids or heparin may be suggested when IL develops following Fontan surgery.
CONCLUSIONS
Until now, there had been no publications regarding this pathology in Colombia and only a few scattered articles in the world literature. In pediatrics, IL must be suspected when an infant develops chronic diarrhea malnutrition, edema, hypoalbuminemia and lymphopenia in the first months of life. Early diagnosis and opportune nutritional treatment address the loss of lymphocytes, immunoglobulins, proteins, fat and micronutrients which can lead to repeated infections and on some occasions to death.
REFERENCES
1. Van der Meer SB, Forget PP, Willebrand D. Intestinal lymphangiectasia without protein loss in a child with abdominal pain. J Pediatr Gastroenterol Nutr 1990; 10: 246-248. [ Links ]
2. Braamskamp M, Dolman K, Tabbers M. Practice protein losing enteropathy in children. Eur J Pediatr 2010; 169:1179-1185. [ Links ]
3. Hoshina T, Kusuhara K, Saito M, Hara T. Cytomegalovirus-associated protein- Losing enteropathy Resulting from lymphangiectasia in a Immunocompetent child. Jpn J Infect Dis 2009; 62(3): 236-8. [ Links ]
4. Hart MH, Vanderhoof JA, Antonson DL. Failure of blind small bowel biopsy in the diagnosis of intestinal lymphangiectasia. J Pediatr Gastroenterol Nutr 1987; 6(5): 803-5. [ Links ]
5. Macdonald J, Porter V, Scott NW, McNamara D. Small bowel lymphangiectasia and angiodysplasia, a positive association; Novel clinical marker or shared pathophysiology? J Clin Gastroenterol Nutr 2010; 44(9): 610-4. [ Links ]
6. Sari S, Baris Z, Dalgic B. Primary intestinal lymphangiectasia in children: Is octreotide an effective and safe option in the treatment? J Pediatr Gastroenterol Nutr 2010; 51(4): 454-7. [ Links ]
7. Uğuralp S, Mutus M, Kutlu O, Cetin S, Baysal T, Mizrak B. Primary intestinal lymphangiectasia: a rare disease in the differential diagnosis of acute abdomen. J Pediatr Gastroenterol Nutr 2001; 33(4): 508-10. [ Links ]
8. Suresh N, Ganesh R, Sankar J, Sathiyasekaran M. Primary intestinal lymphangiectasia. Indian Pediatr 2009; 46(10): 903-6. [ Links ]
9. Dierselhuis MP, Boelens JJ, Versteegh FG, Weemaes C, Wulffraat NM. Recurrent and opportunistic infections in children with primary intestinal lymphangiectasia. J Pediatr Gastroenterol Nutr 2007; 44(3): 382-5. [ Links ]
10. Strober W, Wochner RD, Carbone PP, Waldmann TA. Intestinal lymphangiectasia: a protein-losing enteropathy with hypogammaglobulinemia, lymphocytopenia and impaired homograft rejection. J Clin Invest 1967; 46(10): 1643-56. [ Links ]
11. Tift WL, Lloyd JK. Intestinal lymphangiectasia. Long-term results with MCT diet. Arch Dis Child 1975; 50(4): 269-76. [ Links ]
12. Donzelli F, Norberto L, Marigo A, Barbato A, Tapparello G, Basso G, Zacchello G. Primary intestinal lymphangiectasia. Comparison between endoscopic and radiological findings. Helv Paediat Acta 1980; 35: 169-175. [ Links ]
13. Molina M, Romero A, Antón S, Sarría J, Prieto G, Polanco I. Linfangiectasia intestinal primaria: evolución a largo plazo. An Esp Pediatr 2001; 54(Supl. 3): 33-35. [ Links ]
14. Strygler B, Nicar MJ, Santangelo WC, Porter JL, Fordtran JS. Alpha 1-antitrypsin excretion in stool in normal subjects and in patients with gastrointestinal disorders. Gastroenterology 1990; 99(5): 1380-7. [ Links ]
15. Mine K, Matsubayashi S, Nakai Y, Nakagawa T. Intestinal lymphangiectasia markedly improved with antiplasmin therapy. Gastroenterology 1989; 96(6): 1596-9. [ Links ]
16. Ballinger AB, Farthing MJ. Octreotide in the treatment of intestinal lymphangiectasia. Eur J Gastroenterol Hepatol 1998; 10(8): 699-702. [ Links ]
17. Kuroiwa G, Takayama T, Sato Y, Takahashi Y, Fujita T, Nobuoka A, Kukitsu T, Kato J, Sakamaki S, Niitsu Y. Primary intestinal lymphangiectasia successfully treated with octreotide. J Gastroenterol 2001; 36(2): 129-32. [ Links ]
18. Vardy PA, Lebenthal E, Shwachman H. Intestinal lymphagiectasia: a reappraisal. Pediatrics 1975; 55(6): 842-51. [ Links ]
19. Perez S, Campuzano M, Bousoño C, Ramos P. Ascitis quilosa congénita con linfangiectasia intestinal. Departamento de pediatría. Hospital universitario central de asturias, Oviedo. Bol pediatr 2007; 47: 132-135 [ Links ]
20. Argüelles M, García N, Pavon B. Tratado de gastroenterología, hepatología y nutrición pediátrica aplicada de la SEGHNP. 2011. [ Links ]
