










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.1 Bogotá Jan./Mar. 2014
Enteroscopia total de doble balón intraoperatoria asistida por laparoscopia en el tratamiento de Síndrome de Peutz-Jeghers: reporte de un caso
Rómulo Darío Vargas R., MD (1), Valeria Atenea Costa B. MD (2), Gonzalo Moros, MD (3), Jaime Alvarado Bestene, MD (4)
(1) Internista, Gastroenterólogo, Hospital Universitario San Ignacio. Bogotá, Colombia
(2) Internista, Fellow de Gastroenterología Pontificia Universidad Javeriana, Hospital San Ignacio. Bogotá, Colombia.
(3) Profesor Asociado de Cirugía Laparoscópica y Mínimamente Invasiva. Hospital Universitario San Ignacio. Bogotá, Colombia.
(4)Internista, Gastroenterólogo, Profesor Titular de la Pontificia Universidad Javeriana, Hospital Universitario San Ignacio. Bogotá, Colombia.
Fecha recibido: 16-09-13 Fecha aceptado: 19-12-13
Resumen
El síndrome de Peutz-Jeghers se caracteriza por pólipos hamartomatosos localizados principalmente en el intestino delgado. La mayoría de estos pacientes son sometidos a múltiples resecciones endoscópicas o quirúrgicas. Este caso trata de una paciente femenina quien fue sometida a enteroscopia de doble balón intraoperatoria asistida por laparoscopia para resección de pólipos.
Palabras clave
Síndrome de Peutz-Jeghers, enteroscopia de doble balón, laparoscopia.
INTRODUCCIÓN
Los síndromes de poliposis hamartomatosos constituyen un grupo de al menos seis diferentes enfermedades raras que requieren manejo endoscópico. No hay disponibilidad de estudios prospectivos para probar la validez de los enfoques terapéuticos, pero el conocimiento de las bases genéticas y manifestaciones patológicas de estas enfermedades puede utilizarse para adaptar enfoques de intervención y vigilancia en los pacientes afectados (1).
Los síndromes de poliposis hamartomatosos incluyen la poliposis juvenil, el síndrome de Bannayan–Riley–Ruvalcaba, el síndrome de Peutz–Jeghers, enfermedad de Cowden, y un pequeño número de síndromes de poliposis mixta en los cuales aparecen adenomas, pólipos hiperplásicos y otras lesiones (1).
El síndrome de Peutz–Jeghers es una condición autosómica dominante caracterizada por el desarrollo de pólipos hamartomatosos a lo largo del tracto gastrointestinal y pigmentación mucocutánea asociada (2).
El clásico manejo quirúrgico con múltiples enterotomías y remoción de los pólipos ha sido modificado en años recientes por un enfoque quirúrgico-endoscópico (2). Este enfoque quirúrgico endoscópico realizado en la paciente se explica en la revisión del tema.
REPORTE DE CASO
Paciente mujer de 14 años, con diagnóstico conocido de Síndrome de Peutz–Jeghers desde los 10 años. No hay historia de antecedentes familiares de enfermedad del tracto digestivo.
Al inicio de la enfermedad presentó cuadro de un año de evolución de dolor abdominal intermitente de predominio en epigastrio que aparecía cada 3 meses, posprandial, de aproximadamente 2 horas de duración, el cual mejoraba sin medicación; presentando igualmente episodios de estreñimiento.
Consultó en varias oportunidades por cuadros dolorosos similares con indicación de cambio en la dieta para el estreñimiento e hidróxido de aluminio sin mejoría. En una oportunidad presentó clínica de dolor abdominal, vómitos y pérdida de peso después de cuadro de estreñimiento con impactación fecal que fue manejado con enemas y dieta.
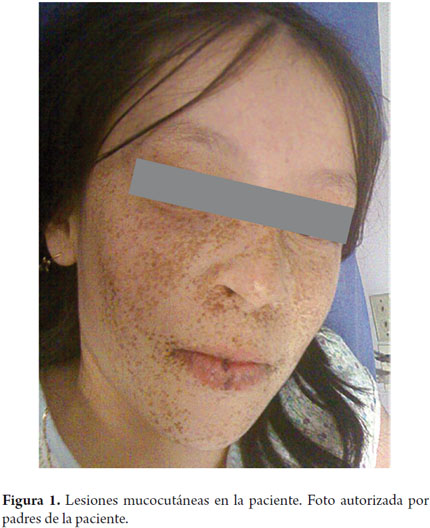
Por persistencia de dolor abdominal se consideró cuadro de obstrucción intestinal. Fue evaluada por gastroenterología pediátrica, sospechando por la presencia de lentiginosis labial y lo anteriormente descrito; un síndrome de Peutz–Jeghers (figura 1).
Se realizó una colonoscopia donde se observó pólipo gigante pediculado de 5 cm en el sigmoides. Se resecó y la biopsia reportó pólipo hamartomatoso con focos de displasia de alto grado y el borde de sección libre de displasia. Se realizó endoscopia de vías digestivas alta, hallándose 20 lesiones polipoides sésiles entre 2 y 4 mm gástricos. Las biopsias de estas lesiones eran compatibles con pólipos hiperplásicos.
Se realizó enteroscopia de doble balón retrógrada observando pólipo sésil diminuto en el colon ascendente, con lesiones nodulares en el íleon terminal. Con base en lo anterior, se formalizó el diagnóstico de Síndrome de Peutz–Jeghers.
En el 2009 se ejecutó la primera laparotomía infraumbilical con resección de intestino delgado por obstrucción intestinal con evolución postoperatoria adecuada. Un mes después se le realizó tránsito intestinal con doble contraste resultando normal.
Presentó otro cuadro de obstrucción intestinal un año luego del diagnóstico de la enfermedad, que ameritó nueva laparotomía. Posteriormente, en el 2011, se diagnosticó intususcepción ileoileal sin abdomen agudo u obstrucción, confirmándose por enteroresonancia, que evidenció un segmento de intestino delgado con imagen en cebolla, dado por la estratificación de las capas del asa dentro de otra asa; sin compromiso isquémico secundario. Se efectuó nueva laparotomía con enterotomía, polipectomía y desinvaginación.
Presentó recurrencia de los pólipos hiperplásicos gástricos sin documentación de Helicobacter pylori y de los pólipos en el intestino delgado resecándose en la última enteroscopia 5 masas polipoides, la mayor de 3,5 cm, cuyas biopsias fueron compatibles con pólipos hamartomatosos sin displasia.
Ingresó al Hospital San Ignacio para polipectomía programada por enteroscopia guiada por laparoscopia. Al examen físico FC: 78 FR: 18, en buen estado general, afebril, hidratada, con máculas hiperpigmentadas en región facial y en labio inferior. Abdomen blando, depresible, no doloroso, heridas quirúrgicas en buen estado.
Se realizó diagnóstico intraoperatorio de intususcepción por laparoscopia diagnóstica más liberación de adherencias, yeyunostomía y enteroscopia operatoria con resección de pólipos. El procedimiento tuvo una duración de 5 horas.
Se exteriorizó un asa intestinal, se realizó jareta con vicryl 3-0 y enterotomía de 2 cm.
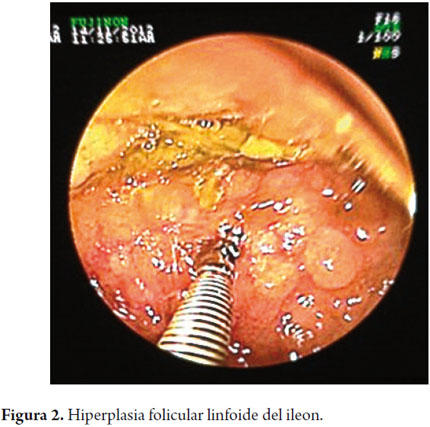
Se insertó el enteroscopio por la boca caudal de la enterotomía y el avance hasta la región ileocecal, se produjo por el desplazamiento de las asas que realizó el cirujano laparoscopista; sobre el enteroscopio a manera de poder plegar el intestino sobre el enteroscopio. Igualmente, el procedimiento se realizó por la boca oral de la enterotomía hasta el ángulo de Treitz. En íleon terminal se observó una mucosa nodular en patrón en empedrado, sugestivo de nódulos linfoides, a este nivel se tomaron biopsias (figura 2). En el íleon terminal, a unos 50 cm de la válvula, se observó un pólipo diminuto de 3 mm que se resecó con asa. En el íleon proximal se observó otra área anastomótica ligeramente irregular, blanda, que se envió a biopsia.
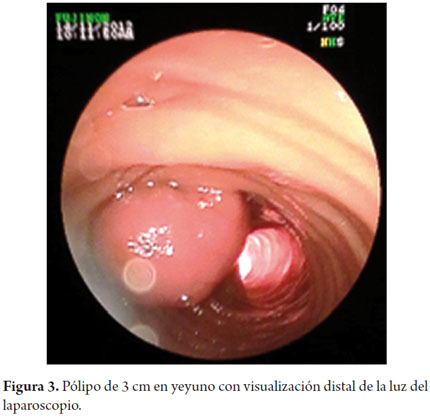
También se halló un pólipo pseudopediculado de 3 cm de diámetro a nivel del yeyuno medio, que correspondía a la cabeza de intususcepción descrita, el cual se resecó por vía endoscópica (figura 3).
Se realizó levantamiento del pólipo con inyección submucosa y se resecó con asa diatérmica en un solo tiempo. Se revisó el área cruenta sin evidenciar perforación, lo cual se verificó por visualización laparoscópica del área.
Se evidenciaron laceraciones de serosa al terminar el procedimiento, a las cuales se les realizó rafias en 3 sitios con vicryl 3-0.
Se realizó resección intestinal del segmento intestinal ostomizado por vía extracorpórea, sección del meso y anastomosis laterolateral con sutura mecánica. Se cerró la enterotomía con técnica de doble grapado y se reforzaron los bordes. Se extrajeron los trocares bajo visión laparoscópica. Se evacuó neumoperitoneo y se cerró la piel.
Dentro de los hallazgos quirúrgicos se evidenció síndrome adherencial viscerovisceral y visceroparietal de predominio infrumbilical. Presentaba múltiples rafias intestinales previas localizadas a 180 cm, 230 y 300 cm del ligamento de Treitz. Igualmente había presencia de área de intususcepción a nivel del yeyuno medio a 180 cm del ligamento de Treitz la cual fue reducida completamente.
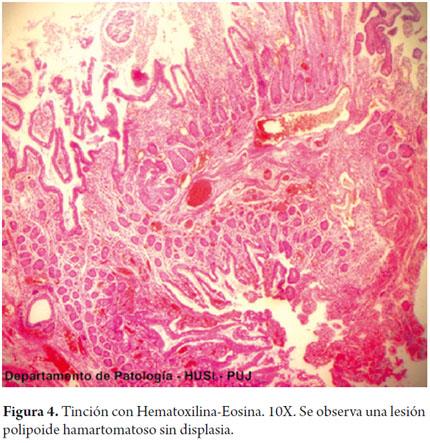
Los diagnósticos anatomopatológicos de los pólipos en yeyuno e íleon distal demostraron pólipos hamartomatosos sin evidencia de cambios displásicos o de malignidad. Igualmente la mucosa de íleon tenía cambios compatibles hiperplasia folicular linfoide (figura 4).
La paciente evolucionó satisfactoriamente en el postoperatorio y no tuvo complicaciones.
DISCUSIÓN
Concepto-diagnóstico
El síndrome fue nombrado por Peutz, luego de observar por primera vez una relación entre los pólipos intestinales y las máculas mucocutáneas en 1921, luego de Jeghers en la década de 1940. El síndrome tiene un cociente de relación hombre–mujer de 1:1, y el promedio de edad en el momento del diagnóstico es de 23 años en los hombres y 26 años en las mujeres (1).
El síndrome de Peutz–Jeghers es un trastorno raro. Es menos común que la poliposis adenomatosa, con una incidencia de 1 en 8300 a 29 000 nacidos vivos (2).
A pesar de que los pólipos hamartomatosos son típicamente lesiones benignas, son los responsables de las mayorías de las complicaciones en el síndrome de Peutz–Jeghers; como la intususcepción, obstrucción intestinal y el sangrado (2).
La distinción de otras poliposis no es problemática generalmente. Los pólipos son más grandes y más numerosos en el intestino delgado, y la condición se presenta típicamente con obstrucción o invaginación intestinal en la segunda o tercera década de vida. El riesgo de intususcepción antes de los 20 años es de 50% (3, 4).
Los criterios diagnósticos del síndrome de Peutz–Jeghers son los siguientes (3):
Dos o más pólipos hamartomatosos
Un pólipo hamartomatoso
Lesiones pigmentarias mucocutáneas
Presencia de un pólipo típico
Historia familiar.
Los pólipos no tienen características endoscópicas específicas y pueden sólo distinguirse de otros tipos de pólipos por histopatología (5).
La historia familiar es negativa en hasta un 45% de los casos. Las mutaciones en el gen LKB1 (STK11) en el cromosoma 19p13 se encuentran en aproximadamente el 50% de familias afectadas. El fenotipo es más severo en las familias con una mutación entera que una mutación sin sentido (2).
Clínica
Las características clínicas predominantes del síndrome de Peutz–Jeghers son el resultado de poliposis gastrointestinal, que pueden conducir a dolor abdominal, intususcepción y sangrado. Los pólipos hamartomatosos pueden detectarse en el 88% de los pacientes con síndrome de Peutz–Jeghers y la mayoría se encuentran en el intestino delgado (6).
El orden de prevalencia es yeyuno, íleon, duodeno, seguido de colon y estómago. Un tercio de los pacientes experimenta síntomas secundarios a pólipos del intestino delgado durante la primera década de vida, y 50% a 60% de los pacientes experimentan síntomas antes de la edad de 20 años, en particular, debido a la obstrucción y la intususcepción intestinal (6).
Histología
Las células representadas en un hamartoma típicamente derivan de progenitores estromales o mesenquimales, aunque también pueden estar involucrados elementos ectodérmicos o endodérmicos también (5).
Los pólipos en su mayoría son hamartomas, pero en algunos casos los pólipos tienen cambios adenomatosos y focos de adenocarcinoma (7).
La displasia es poco común, aunque se ha sugerido una secuencia hamartoma-adenoma-carcinoma (2, 4).
Los pólipos son típicamente multilobulados con una superficie papilar y se asemejan a los adenomas tubulovellosos. Estos pólipos están cubiertos por epitelio hiperplásico. Puede haber desplazamiento del epitelio secretor de mucina en la submucosa, muscularis propia y más allá de la pared del intestino, causando mímica con el adenocarcinoma bien diferenciado o mucinoso (2).
Los pólipos hamartomatosos tienen características histológicas típicas, consistente en la presencia de músculo liso ramificado, tipo árbol cubierto de epitelio normal (3).
Riesgo de cáncer
Los pacientes que padecen este síndrome tienen un mayor riesgo de cáncer y patologías malignas extracolónicas. Los principales sitios de tumores extracolónicos son el páncreas, estómago, mama, ovario, testículo (tumores de la célula de Sertoli) y cuello uterino (4).
El riesgo de desarrollar cáncer es de 85% a la edad de 70 años. Un estudio que incluía 419 pacientes informó un riesgo de cáncer gastrointestinal de 57% a los 70 años. El cáncer colorrectal fue el cáncer más común con un riesgo de vida de 39%, mientras que el riesgo acumulado de cáncer pancreático fue 11% (4).
También se aumenta el riesgo de cáncer gástrico, intestino delgado y biliar. El riesgo reportado de desarrollar cáncer de mama del 31% al 50% (4).
Giardiello ha proporcionado el mejor análisis del riesgo del cáncer en el síndrome de Peutz–Jeghers mediante el estudio de 210 pacientes. El riesgo relativo para cualquier cáncer durante la vida de estos pacientes es de 15,2, con riesgos en todas las porciones del intestino, pulmones, mama, útero y gónadas. El riesgo acumulado para cualquier cáncer entre los 15 a 64 años de edad es de un valor asombroso de 93% (1).
Tratamiento
Algunos investigadores han sugerido que los pólipos mayores de 1,5 cm se deben quitar en la detección. Sin embargo, en un estudio de Taiwan, algunos pacientes que se sometieron a cirugía abdominal para la extirpación de pólipos, experimentaron complicaciones postquirúrgicas como el síndrome del intestino corto e íleo, dando lugar a un estado nutricional deficiente y mala calidad de vida (7).
Clásicamente las complicaciones secundarias a la poliposis del intestino delgado fueron manejadas con laparotomías exploradoras con reducción de áreas de intususcepción, polipectomías quirúrgicas a través de múltiples enterotomías y resecciones segmentarias del intestino delgado. La palpación externa y la transiluminación eran métodos convencionales para la identificación de los pólipos durante el acto quirúrgico, siendo inadecuados para la identificación de pólipos pequeños conllevando a repetir laparotomías y resecciones adicionales del intestino delgado. Esto exponía a los pacientes a mayor morbimortalidad relacionada con los múltiples procedimientos quirúrgicos y ocasionalmente se complicaban con síndrome de intestino corto (2).
Se ha descrito que el seguimiento en estos pacientes debe ser en intervalos de 2 años, consistente en radiografías contrastadas seguido de enteroscopia intraoperatoria para la remoción de pólipos mayores de 15 mm (8).
El valor diagnóstico de la videocápsula endoscópica ha sido estudiado en pacientes con síndrome de Peutz–Jeghers. En general es segura, bien tolerada y valiosa para la detección de pólipos en el intestino delgado. Aunque la video cápsula es capaz de detectar pólipos menores de 5 mm, no puede determinar con exactitud la ubicación y el tamaño exacto de los pólipos. Además, con frecuencia se divulgan resultados falsos negativos de la videocápsula y hay evidencia creciente en ciertos estudios de que incluso las grandes lesiones pueden ser ignoradas. Un estudio portugués informó un 20% (5/26) de los pólipos mayores de 11 mm no fueron diagnosticados por videocápsula en 14 pacientes (3).
Ya que las modalidades de imágenes radiográficas y videocápsula endoscópica son pruebas puramente diagnósticas, las opciones terapéuticas para pólipos identificados del intestino delgado han sido limitadas a la enteroscopia de doble balón y enteroscopia asistida por laparoscopia. La primera es la menos invasiva de estas opciones terapéuticas, porque puede realizarse de forma ambulatoria, sin necesidad de asistencia quirúrgica. La formación de asa puede limitar el control endoscópico para la resección de pólipos grandes (8).
Dentro de la literatura, la enteroscopia de doble balón se ha realizado con éxito en niños tan jóvenes como de 2 años de edad. Hay que tener en cuenta que la cavidad abdominal es más pequeña, más delgadas las paredes intestinales y más estrecho el lumen intestinal. En los niños más pequeños en comparación con los adultos es técnicamente más difícil y probablemente requiere un mayor nivel de habilidad para el desempeño exitoso (9).
Muchos pacientes con síndrome de Peutz–Jeghers han tenido cirugías abdominales previas con posterior formación de adherencias intraabdominales. Debido a que la técnica de enteroscopia de doble balón requiere libre movimiento del intestino dentro de la cavidad abdominal, la misma se ve limitada por la formación de adherencias intraabdominales post quirúrgicas (8).
El clásico manejo quirúrgico con múltiples enterotomías y remoción de los pólipos ha sido modificado en años recientes por un enfoque quirúrgico-endoscópico (2).
Ohmiya y colaboradores fueron los primeros en describir el uso de enteroscopia de doble balón para el diagnóstico y tratamiento del síndrome de Peutz–Jeghers. En dos pacientes, con pólipos múltiples entre 10-60 mm detectados en el yeyuno y el íleon, estos fueron resecados sin sangrado posterior o perforación. Desde entonces, varios estudios han descrito un alto rendimiento diagnóstico y terapéutico de la enteroscopia de doble balón en pacientes con síndrome de Peutz–Jeghers (3).
Yamamoto y Sugano desarrollaron una técnica de inserción de enteroscopia de doble balón, para mejorar el acceso al intestino delgado dentro de un período relativamente corto de tiempo. El método de doble balón permite no sólo la visualización endoscópica de la totalidad del intestino delgado, sino también muestras de tejido y terapias intervencionistas, incluyendo la dilatación con balón de estenosis benignas y polipectomía (10).
Las obstrucciones del intestino delgado en paciente con síndrome de Peutz–Jeghers se pueden manejar exitosamente con abordajes mínimamente invasivos. El tratamiento de la obstrucción en estos pacientes es eliminar el pólipo hamartomatoso complicado. La repetición de episodios de intususcepción intestinal ocurre en al menos el 10% de los casos. Esto puede hacerse intraoperatoriamente con colonoscopia y enteroscopia asistida laparoscópicamente. Una vez que se han eliminado los pólipos del tracto gastrointestinal, el intervalo recomendado de seguimiento del intestino delgado es de 2 a 3 años. La presencia de pólipos mayores de 1,5 cm en diámetro exige otra evaluación gastrointestinal completa con extracción endoscópica de los pólipos (10).
En general, la tasa de complicaciones después de la enteroscopia de doble balón se estima en 0,8%; la más grave es la pancreatitis aguda con un riesgo de aproximadamente 0,2% a 0,3% después de abordaje anterógrado (3).
Como en la endoscopia convencional, el riesgo de complicaciones graves es mayor en enteroscopia terapéutica (4,3%), principalmente atribuible al sangrado (3%) (3).
Por lo tanto, el uso de enteroscopia de doble balón en forma serial podría proporcionar un medio para realización de polipectomías profilácticas en pacientes con síndrome de Peutz–Jegher, para así prevenir complicaciones como la invaginación intestinal y el sangrado (11).
Una institución japonesa desarrolló la siguiente estrategia terapéutica para los pacientes con síndrome de Peutz–Jeghers. La resección endoscópica de los pólipos del intestino delgado superiores a 20 mm fue priorizada para reducir el riesgo de invaginación intestinal. Sólo se recuperaron pólipos grandes o de forma irregular para examinación histopatológica. La mayoría de los pólipos pequeños fueron resecados sin recuperación (11).
Si todos los pólipos grandes no pudieron ser resecados en la primera sesión, la próxima sesión fue programada dentro de 6 meses. El seguimiento endoscópico fue realizado a intervalos regulares (anual) después de que todos los pólipos grandes habían sido resecados (11).
En este estudio, se evaluó la eficacia de esta estrategia en el manejo endoscópico de los pólipos del intestino delgado en pacientes con síndrome de Peutz–Jeghers (11).
Se incluyeron 15 pacientes entre los años 2000 y 2009. Un total de 88 enteroscopias de doble balón fueron realizadas en estos pacientes durante el periodo mencionado. El número promedio de enteroscopias de doble balón por paciente fue 5,9, con rango entre 3-10 (11).
La enteroscopia de doble balón permite una alta tasa de éxito para la inspección de la totalidad del intestino delgado. Las tasas de éxito de enteroscopia total se conocen entre 40% a 80%. La tasa de éxito para la enteroscopia total en este estudio fue 70,0% (11).
La técnica de enteroscopia intraoperatoria varía en varios aspectos importantes, tales como el enfoque de acceso intraabdominal (laparotomía versus laparoscopia), el endoscopio utilizado y la técnica de inserción del endoscopio (12).
El procedimiento estándar consiste en una laparotomía seguida de enterotomía (generalmente en el intestino medio) o dos pequeñas incisiones a través de las cuales se introduce el endoscopio (un colonoscopio estándar, preferiblemente pediátrico, un enteroscopio de pulsión o incluso un gastroscopio estándar) (12).
scopia intraoperatoria a través de una enterotomía ofrece la mejor opción para examinar el intestino delgado en su totalidad y para disminuir el trauma (12).La enteroscopia intraoperatoria puede realizarse mediante laparotomía o por vía asistida por laparoscopia. Aunque estos enfoques permiten un sólo tiempo, la inspección completa del intestino y la resección de los pólipos, la manipulación quirúrgica del intestino puede ser significativa y llevar a dilatación intestinal intraoperatoria además de íleo postoperatorio importante (8).
La técnica combinada endoscópica quirúrgica fue inicialmente descrita en 1985 por Mathus, Vliegen y Tytgat cuando trataron 5 casos con 5 a 20 polipectomías individuales. Esto fue seguido de varios reportes del uso de técnicas similares para abordar intraoperatoriamente el intestino delgado. El uso de la técnica combinada provee un intestino delgado limpio con máximo control de la poliposis. Este abordaje disminuye la tasa de laparotomías y el síndrome de intestino corto, y alarga el periodo asintomático en estos pacientes. Igualmente la excisión de pequeños pólipos que pueden pasar desapercibidos cuando se realiza solo cirugía (2).
Ha sido descrita la reducción laparoscópica en pacientes con poliposis complicada con intususcepción, también hay casos de enteroscopia asistida laparoscópicamente en pacientes con sangrado intestinal (2, 10, 13, 14).
Al combinar la enteroscopia de doble balón con la laparoscopia, se ha desarrollado un procedimiento mínimamente invasivo, en un solo paso, que puede utilizarse para el seguimiento de pólipos del intestino delgado y para tratamiento en los pacientes con síndrome de Peutz–Jeghers (8).
Complicaciones
Las tasas de pancreatitis aguda, hemorragia tardía, perforación y la totalidad de complicaciones en pacientes que experimentaron enteroscopia de doble balón terapéuticas fueron 2,7%, 2,7%, 1,4% y 6.8%, respectivamente (11).
Las tasas de complicaciones de enteroscopia intraoperatoria han oscilado entre 0% a 52% e incluyen hematomas intramurales, laceración de la mucosa, perforación, hemorragia mesentérica, íleo paralítico, pancreatitis, isquemia intestinal, obstrucción intestinal, infección de la herida e infecciones pulmonares postoperatoria (12).
Autores holandeses describieron 29 procedimientos diagnósticos y terapéuticos de enteroscopia de doble balón en 13 pacientes con síndrome de Peutz–Jeghers con polipectomías múltiples de pólipos de 10 mm. Ninguna complicación ocurrió durante los procedimientos ni durante el seguimiento. Sin embargo, otros dos estudios informan una tasa de complicaciones de hasta 6,8%, incluyendo la pancreatitis aguda (2,7%) y el síndrome pospolipectomía (5%) (3).
Un estudio evaluó la viabilidad y utilidad de la enteroscopia de doble balón en el manejo de enfermedades del intestino en los niños (edad promedio de 12,9 años), incluyendo 5 pacientes conocidos con síndrome de Peutz–Jeghers. En 2 de los 5 pacientes, la enteroscopia completa fue alcanzada y la polipectomía fue exitosa. No hubo complicaciones relacionadas con la enteroscopia, sin embargo, un paciente al que se le realizo enteroscopia de doble balón asistida por laparoscopia desarrolló un absceso pélvico sin perforación intestinal, probablemente como complicación de la laparoscopia (3).
Aunque la mayoría de los estudios no informan mortalidad, la mortalidad relacionada con el procedimiento o con complicaciones postoperatorias ha sido reportada en un 11%. En un estudio, un paciente murió de peritonitis después de la resección del intestino delgado y del desarrollo de una fístula anastomótica (12).
Consideramos importante reportar este caso, ya que en la búsqueda de literatura con respecto al tema, en Colombia, no han sido reportados casos de pacientes con síndrome de Peutz–Jeghers a quienes se les haya realizado polipectomía electiva por enteroscopia de doble balón guiada por laparoscopia.
La finalidad de presentar el caso, está en relación a mostrar la relevancia en el manejo multidisciplinario de una paciente mínimamente abordada, a pesar de múltiples reintervenciones previas.
Fue técnicamente posible, mostrándose como una técnica segura, reproducible y con excelentes resultados.
REFERENCIAS
1. Shin SK, Boland CR. Endoscopic Management in Juvenile Polyposis, Peutz-Jeghers Syndrome, and Other Hamartomatous Polyposis Syndromes. Techniques in Gastrointestinal Endoscopy. 2006;8(3):119-25. [ Links ]
2. Amaro R, Diaz G, Schneider J, Hellinger MD, Stollman NH. Peutz-Jeghers syndrome managed with a complete intraoperative endoscopy and extensive polypectomy. Gastrointest Endosc. 2000;52(4):552-4. [ Links ]
3. Korsse SE, Dewint P, Kuipers EJ, van Leerdam ME. Small bowel endoscopy and Peutz-Jeghers syndrome. Best Pract Res Clin Gastroenterol. 2012;26(3):263-78. [ Links ]
4. Jass JR. Gastrointestinal polyposes: clinical, pathological and molecular features. Gastroenterol Clin North Am. 2007;36(4):927-946, viii. [ Links ]
5. Kopacova M, Tacheci I, Rejchrt S, Bures J. Peutz-Jeghers syndrome: diagnostic and therapeutic approach. World J Gastroenterol. 2009;15(43):5397-408. [ Links ]
6. Ohmiya N, Taguchi A, Shirai K, Mabuchi N, Arakawa D, Kanazawa H, et al. Endoscopic resection of Peutz-Jeghers polyps throughout the small intestine at double-balloon enteroscopy without laparotomy. Gastrointest Endosc. 2005;61(1):140-7. [ Links ]
7. Chen T-H, Lin W-P, Su M-Y, Hsu C-M, Chiu C-T, Chen P-C, et al. Balloon-assisted enteroscopy with prophylactic polypectomy for Peutz-Jeghers syndrome: experience in Taiwan. Dig Dis Sci. 2011;56(5):1472-5. [ Links ].
8. Ross AS, Dye C, Prachand VN. Laparoscopic-assisted double-balloon enteroscopy for small-bowel polyp surveillance and treatment in patients with Peutz-Jeghers syndrome. Gastrointest Endosc. 2006;64(6):984-8. [ Links ].
9. Lin TK. Enteroscopy in the pediatric population. Techniques in Gastrointestinal Endoscopy. 2013;15(1):36-40. [ Links ]
10. Gonzalez AMM, Clapp B. Laparoscopic management of small bowel intussusception in a 16-year-old with Peutz-Jeghers syndrome. JSLS. 2008;12(3):332-4. [ Links ]
11. Sakamoto H, Yamamoto H, Hayashi Y, Yano T, Miyata T, Nishimura N, et al. Nonsurgical management of small-bowel polyps in Peutz-Jeghers syndrome with extensive polypectomy by using double-balloon endoscopy. Gastrointest Endosc. 2011;74(2):328-33. [ Links ]
12. Schulz H-J, Schmidt H. Intraoperative enteroscopy. Gastrointest Endosc Clin N Am. 2009;19(3):371-9. [ Links ]
13. Zanoni ECA, Averbach M, Borges JLA, Corrêa PAFP, Cutait R. Laparoscopic treatment of intestinal intussusception in the Peutz-Jeghers syndrome: case report and review of the literature. Surg Laparosc Endosc Percutan Tech. 2003;13(4):280-2. [ Links ]
14. Kong S-S, Taib NA, Mahadeva S. Successful management of intussusception with total polyp clearance in Peutz-Jeghers syndrome using a combined endoscopic and surgical approach. BMJ Case Rep. 2009;2009. [ Links ]
