










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.2 Bogotá Apr./June 2014
Approaches to Pathological Diagnosis of Cholestatic Diseases
Rocío del Pilar López Panqueva MD. (1)
(1) Pathologist at the Hospital Universitario Fundación Santa Fe de Bogotá and the Universidad de Los Andes in Bogotá, Colombia.
Received: 12-05-14 Accepted: 19-05-14
Abstract
Any disease that leads to impaired bile flow or impaired bile salt metabolism results in cholestasis. There are several causes of the disease related to intrahepatic or extrahepatic anatomical locations, to whether the disease is acute or chronic, to whether or not hepatocellular damage occurs, and to whether or not the condition is primary or secondary. The large number of entities that must be considered in the differential diagnosis of cholestatic diseases poses a major diagnostic challenge for both the clinician and the pathologist (1). This article establishes a diagnostic approach based on histologic patterns which emphasizes adult chronic cholestatic diseases. The next article will focus on the pediatric population.
Keywords
Cholestasis, acute cholestasis, chronic cholestasis, liver biopsy, acute cholangitis, primary biliary cirrhosis, primary sclerosing cholangitis, IgG4 cholangitis.
INTRODUCTION
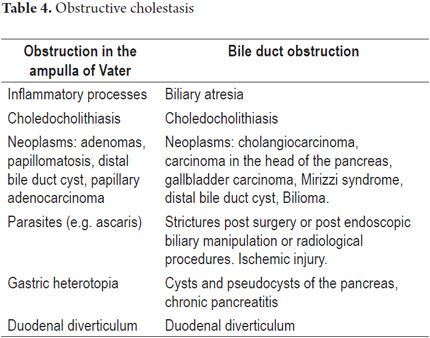
It is important to remember that the liver uses active transport for secretion of bile salts from the canaliculi and through the hepatocellular slits. After being secreted, they attract the remaining components of the bile, especially sodium and water, by osmosis. This bile flow passes through a system of collection ducts. These are bile ducts that drain the bile in a circuit that surrounds the hepatocytes. They then form intrahepatic bile ducts until they reach the right and left hepatic ducts. This is the common bile duct that together with the cystic duct drains the bile stored in the gallbladder. They also constitute the choledochal ducts that lead into the second portion of the duodenum at the ampulla of Vater. These the bile solubilizes fats and vitamins helping with digestion and absorption of nutrients. Cholestasis may occur because of an injury to any of these regions: it is classified into intrahepatic and extrahepatic cholestasis. Tables 1 and 2 summarize some of the possible etiologies. They occur with or without hepatocellular damage and may be acute or recurrent. If the condition persists for more than 6 months, it is called chronic cholestasis (Tables 3 and 4). Cholestasis may occur during both adulthood and childhood.


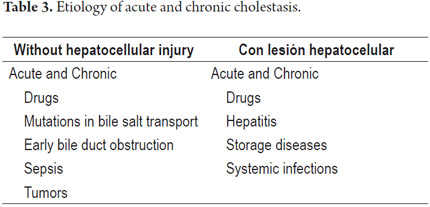
The vast majority of cases of extrahepatic cholestasis can be resolved endoscopically, with radiological treatment or with invasive procedures, but intrahepatic cholestasis usually requires other kinds of therapies (Table 5). Clinical manifestations result from the accumulation of bile in the parenchyma of the liver, in the blood and in tissues into which substances that physiologically contain bile can be excreted. Thus, sometimes metabolic dysfunction of bile acids is indirectly related to accompanying alterations in bilirubin metabolism. This results in jaundice of the skin and/or mucous membranes accompanied by choluria, acholia and hypocholia. Pruritus is a cardinal symptom in chronic cholestatic diseases. Other symptoms are manifestations resulting from bad absorption such as steatorrhea, deficient absorption of liposoluble vitamins, xanthomas, xanthelasma, coagulation disorders, osteopenia, osteoporosis and osteomalacia. Some of the biochemical alterations observed are hyperbilirubinemia, increased alkaline phosphatase, increased gamma-glutamyl, hypercholesterolemia and increased aminotransferases. We must always check for viral diseases, especially hepatitis C and B, and rule them out before making a diagnosis. We also need the patient's medical history regarding metabolic syndrome and intake of toxins such as alcohol or drugs. Diagnostic imaging may also help in the differential diagnosis of extra or intra hepatic cholestasis or even pinpoint the etiological diagnosis. However, it is sometimes necessary to perform a liver biopsy to determine the etiology of cholestasis. It may also be necessary to check other parameters such as inflammatory activity and structural changes and linked pathologies and to establish whether there is progression of an already diagnosed pathology. This article emphasizes the most important and useful histopathological findings for especially for adult and infant populations. It emphasized performance of a joint analysis including a complete medical history and laboratory and imaging studies (2, 3, 4).
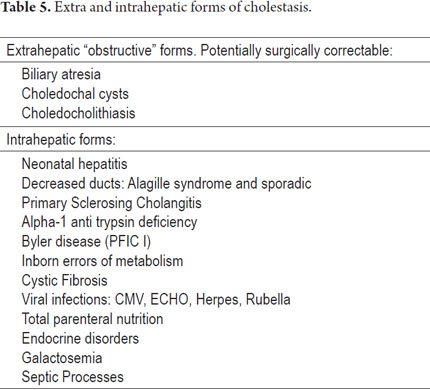
HOW TO ASSESS CHOLESTATIC DISEASE HISTOPATHOLOGICALLY
In the earlier stages of cholestasis, changes are absolutely reversible and ultrastructural. Edema and periportal hepatocytes (hydropic change) can be observed together with reinforcement of the cytoplasmic membrane, mitochondrial edema, megamitochondria and hypertrophy of the rough and smooth endoplasmic reticulum. As the lesion persists there may be noticeable microscopic changes.
The morphological changes we see in histopathological studies are divided into those that are common to the majority of pathologies most diseases. Short physiopathological explanations and diagnostic approaches based on morphological patterns for each pathology are presented below. Finally, this article presents the diagnostic clues most frequently in pathological studies.
We will see then, common observed changes in almost all acute and chronic cholestatic diseases which are presented at the hepatocellular, canalicular and ductal levels.
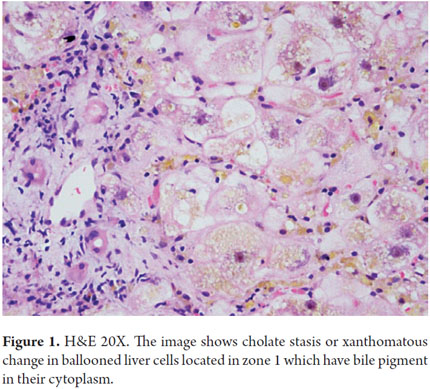
Hepatocellular damage is caused by the presence of bile salts retained within hepatocytes. One of the first symptoms of hepatocellular damage is edema which gives an image known as "cholate stasis". This is a xanthomatous change located in the periportal or peri-septal areas which gives a clearer look at this area. We also observe Mallory-Denk bodies, copper deposits and/or copper binding proteins (Figures 1 and 2).
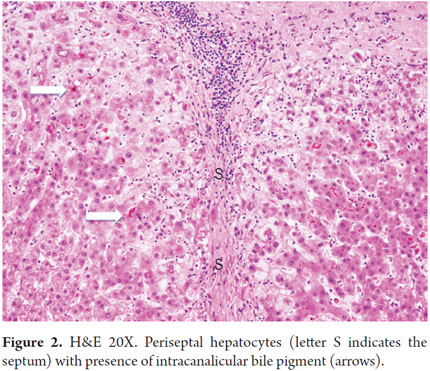
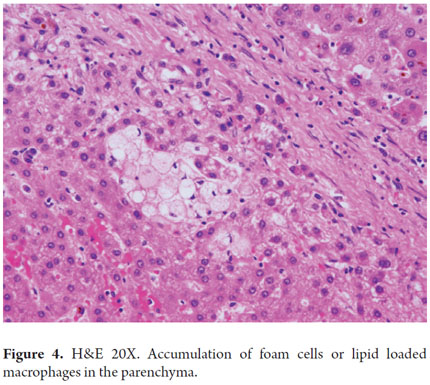
Biliary stasis and loss of the tone of the pericanalicular microfilaments damage the canalicular membrane. This leads to damage of intercellular junctions that allows bile to enter the blood. This leads to bilirubinostasis that is first canalicular and then ductal. If the condition persists, it can lead to the formation of bile plugs (Figure 3) with serious duct or canalicular dilation accompanied by inflammation and accumulation of macrophages filled with lipids at the parenchyma (Figure 4).
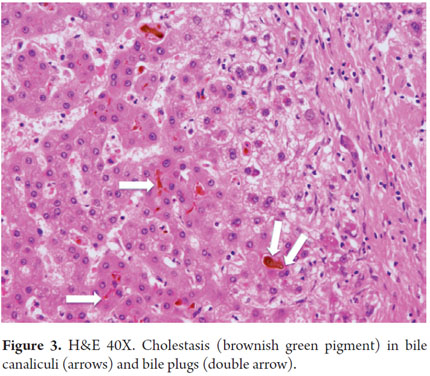
Histochemical studies are very useful for determining whether differences in the brown pigment, sometimes very subtle differences, observed in the hepatocytes, really correspond to bile pigment. For instance, Prussian blue identifies iron deposits while Ziehl-Nielsen stain differentiates liposfuscin pigment (positive staining) from bile pigment (negative staining) which can look very similar when routine stains are used.
In an attempt to help improve the bile flow secondary bile canaliculi proliferate into a maze of small angled irregular ducts arranged in the periphery of the portal tracts. This ductular reaction is derived from periportal hepatic stem cell (HPSCs) that were first described a decade ago. This process reflects the interaction of autocrine and paracrine mediators that includes the release of cytokines such as interleukin 8 (5, 6).
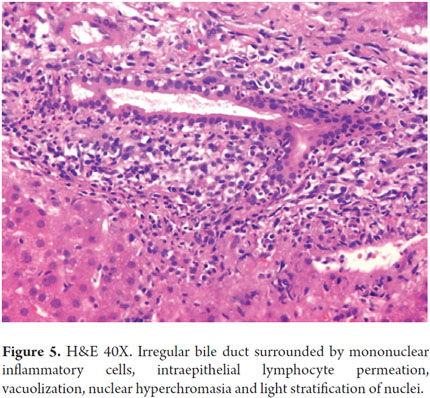
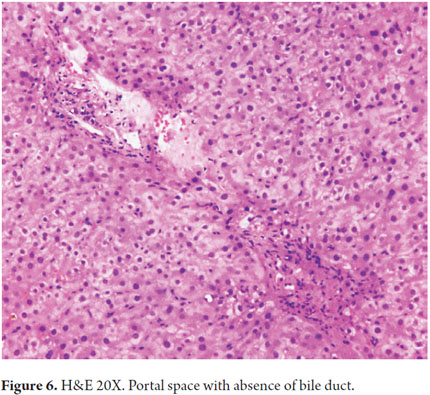
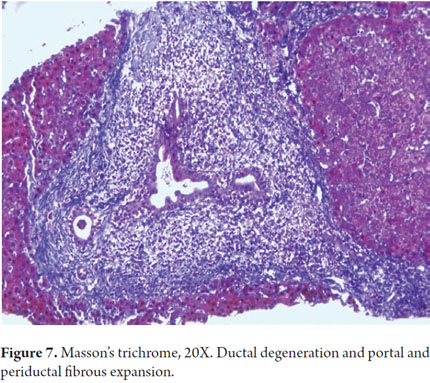
Portal inflammation especially compromises the bile ducts with inflammatory cells permeating the epithelium. Degenerative changes occur including contour irregularity, nuclear pseudostratification, hyperchromasia, and cell vacuolization. Occasionally, mitosis is observed (Figure 5). It is also essential to assess loss of bile ducts. It is considered that under normal conditions up to 7% of the hepatic arteries are not accompanied by bile ducts, but if more than 50% of the portal bile ducts are absent with only the presence of their vascular structures, it is considered true ducts loss or ductopenia (Figure 6). Sometimes, in order to corroborate this, it is useful to perform immunohistochemical studies of cytokeratin 7 or 19 with which the bile ducts become evident when they are not clearly observed with hematoxylin eosin or histochemistry. Later, when fibrosis starts, collagen proliferation widens the portal spaces and periductal and periductular sclerosis develops (Figure 7).
With this understanding of the general histopathological changes, and following the recommendations of Dr Jay H. Lefkowitch, we will describe the diagnoses of some of the more common cholestatic diseases based on morphological patterns (7, 8).
The histologic pattern of "acute cholestasis" refers to occurrences that last from one day to a few days or weeks during which time we can observe four different patterns.
OBSTRUCTIVE CHOLESTASIS
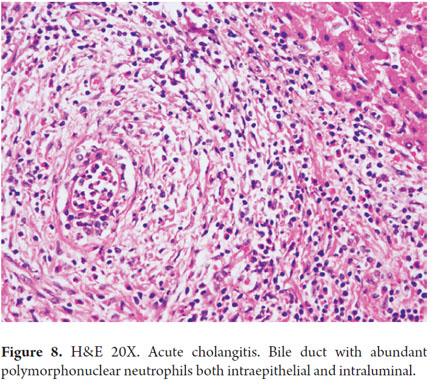
Obstructive cholestasis is caused by a mechanical impairment of bile flow through large ducts including extrahepatic bile ducts, or through the perihilar ducts. We summarize some etiological examples in Table 4. It is morphologically translated into parenchymal cholestasis with biliary stasis located in zone 3 (perivenular). Three is bile pigment in hepatocytes and canaliculi, and sometimes there are Kupffer cells (Figure 7). Depending on the site of obstruction, it may compromise bile ducts, focal points in the liver, several liver zones or zones compromising large areas of the parenchyma. These indications are in addition to the classic the portal triad including edema, ductular proliferation, inflammatory infiltrate (predominantly polymorphonuclear neutrophils) and acute cholangitis (Figure 8). Sometimes it is mixed with lymphocytes or plasma cells as described in 1970 by Poulsen (9).
Bile secretion caused by residual hepatocyte function usually persists, and bile circulates due to reabsorption by the capillaries at the tight intercellular junctions specifically in the periportal area. If the obstruction is incomplete, a ductular reaction without evidence of cholestasis can occur. This happens when the obstruction is segmented, the perihilar ducts are intact, or when a biliary stent has recently been placed.
Pure or soft cholestasis
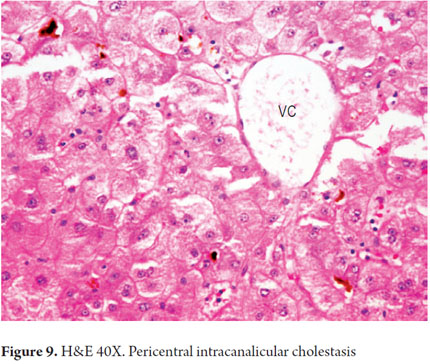
The characteristic feature of this type of cholestasis is the presence of bile inside the hepatocytes and/or bile canaliculi of the centrilobular region or zone 3 without significant portal or parenchymal alterations (Figure 9).
This pattern is characteristic of intrahepatic cholestasis observed in septic processes (caused by cytokines and endotoxins circulation) and in some cases of drug toxicity when functional bile flow impairment is found in lesion perfusion/reperfusion during liver transplant. It is also associated with hypoxia, ischemia, and mutations in the bile salt transport proteins. This occurs for example in progressive familial intrahepatic cholestasis, intrahepatic benign recurrent cholestasis and in some mitochondriopathies (10, 11, 12, 13).
Intrahepatic bile duct diseases
The presence of interlobular bile ducts in the hepatic biopsy accompanied by arterioles of similar caliber are absolutely necessary for identification of this pattern.
The classic example is Primary Biliary Cirrhosis (PBC). In its early stages we find bile duct damage secondary to inflammation of the epithelial cells of the interlobular ducts. This occurs together with cell marginalization of the intraepithelial lymphocytes, plasma cells and/or some eosinophils and leads to degenerative changes of the ductal epithelium with apoptosis, vacuolization and nuclear pseudostratification which are characteristic of this condition (Figure 10) (14).
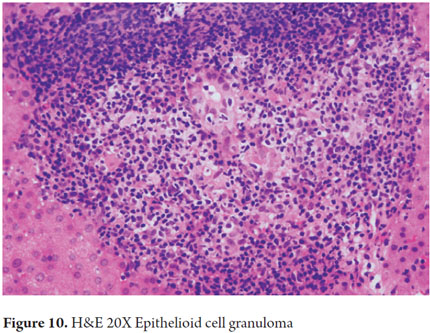
Very similar changes occur in secondary duct lesions due to drug toxicity such as amoxicillin and in acute cell rejection following a liver transplant. Ductopenic diseases are also included on this pattern. There is loss of at least 50% of the bile duct portals. These diseases include "vanishing bile duct syndrome", Allagille syndrome, idiopathic adult ductopenia and classic chronic cholestatic diseases such as primary biliary cirrhosis and primary sclerosing cholangitis.
Hepatitis associated with cholestasis
In this pattern we observe the classical changes of hepatitis with lobular disarray, ballooning, apoptosis, lobular and portal inflammation. Cholestasis can be either inconspicuous or mild, as in those described in viral hepatitis B and C. It is much more frequently observed in HVA and HVE. It can also be observed in some cases of alcoholic hepatitis, in drug-induced hepatitis and in autoimmune hepatitis.
Histological patterns of chronic cholestasis
This pattern develops as the result of prolonged cholestasis lasting for several weeks, months or for even longer periods. Large bile ducts are compromised and damaged including even the intrahepatic bile ducts similar to primary biliary cirrhosis (PBC) and intrahepatic and extrahepatic primary sclerosing cholangitis (PSC). These conditions may appear to be very similar in later stages.
Cholestasis progresses from centrilobular to panlobular with xanthomatous changes or cholate stasis in periportal and/or periseptal hepatocytes. There are copper deposits, and copper binding proteins Mallory-Denk bodies are present. The progression of these changes leads to biliary cirrhosis with irregular nodules and a halo effect which corresponds to clear appearance of the hepatocytes in the nodular periphery. This occurs as the result of the mixture of periseptal edema and pseudoxanthomatous change.
Type 1 pattern: the presence of classical portal and/or periportal fibrosis
We observe Type 1 mainly when chronic obstruction of a large bile duct leads to expansion with portal fibrosis and portal to portal bridging. It is accompanied by ductular proliferation although the interlobular bile ducts remain intact.
Typical examples are obstruction of large bile ducts by a tumor in the pancreatic head such as adenocarcinoma of the pancreatic head, biliary stricture and atresia of the extrahepatic bile duct.
Type 2 pattern: periductal fibrosis with ductal abnormalities
The lesion is centered in the portal bile ducts as in primary sclerosing cholangitis with periductal fibro-obliterating lesions forming images of onion rings in both small and large ducts. Lesions Similar to those in other obstructive processes are also seen in secondary sclerosing cholangitis.
Type 3 pattern: ductopenia and atypical ductal destruction
In chronic cholestatic diseases there is duct loss with or without ductular reaction and fibrosis which is very similar to Type 1. Lymphoid aggregates can also exist. Examples of these are PBC, PSC and toxic drug reactions.
We will now explain diagnostic clues from the purely pathological point of view of the most famous adult disorders.
PRIMARY BILIARY CIRRHOSIS (PBC)
Primary Biliary Cirrhosis is considered to be an idiopathic autoimmune disease that most often attacks middle aged women. The female to male ratio is 9-10:1. Before 30 years of ageit is rare and occurs with patient histories of other autoimmune diseases such as thyroiditis, Sjogren's syndrome, rheumatoid arthritis, Raynaud's syndrome, keratoconjunctivitis, and generalized compromise of the ductal epithelium of the salivary, lacrimal and pancreatic biliary glands.
Diagnosis is based on the combination of 3 criteria: biochemical evidence of cholestasis especially increased levels of alkaline phosphatase for at least 6 months; presence of antimitochondrial antibodies (AMA) in titers of more than 1:40 (present in 90-95% of patients); and histopathological evidence of non-suppurative cholangitis with immune-mediated destruction of small and medium caliber bile ducts. The expert consensus recommends that a diagnosis be based on the presence of at least 2 of these three criteria (Evidence Class I level B) (15, 16, 17).
The main problem for histopathological diagnosis is viewing the ductal lesions. Thus, if possible, it is advisable to obtain adequate samples of both the right and left liver, to have at least 10 to 15 portal spaces, to perform serial cut sections of hematoxylin & eosin and to use complete histochemical stains including for copper and copper binding protein. It is also clear that the reasons that lead the clinician to perform a liver biopsy are limited. It can be useful to establish a diagnosis in patients who are AMA negative, to confirm the diagnosis even with positive AMA, to exclude concurrent diseases such as steatohepatitis or overlap syndromes (PBC plus autoimmune hepatitis or PBC with primary sclerosing cholangitis), to stage and to monitor disease progression or response to treatment (17).
In the early stages, changes are focused within the portal tracts. As the disease progresses we observe parenchymal effects of chronic cholestasis. Even the inflammation can cross the limiting plate, although it is minimal. In addition, it is difficult to distinguish PBC from chronic hepatitis and from necroinflammatory disease.
What should be taken into account?
- Non-suppurative cholangitis, ductal destructive, characterized by inflammatory infiltrate especially of plasma cells, with lymphocytes, lack of eosinophils and macrophages in portal locations.
- Florid duct lesions are indicated by three components: inflammation, ductal epithelial damage and disruption of the basal membrane of the bile duct. Interlobular bile ducts ranging from 40 to 80 μ in diameter are typically those which are compromised.
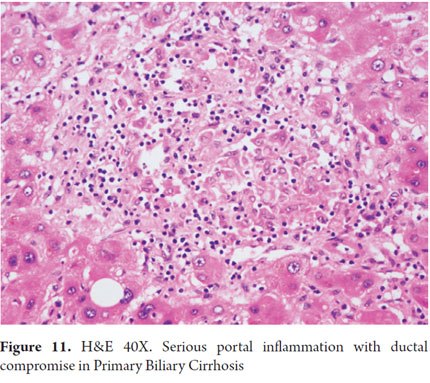
- Granulomatous cholangitis is the presence of granulomas composed of epithelioid cells which are not necrotizing. They are usually poorly formed or pseudogranulomas and are most often found in the portal tracts or surrounding the bile ducts that show degenerative changes (Figure 11). Occasionally there are granulomas and accumulation of lobular macrophages. Granuloma formation results from a complex interaction of activated macrophages and potent chemotactic mediators that capture and perform differentiation, proliferation and accumulation of monocytes and T cells in the portal tract (18).
- In later stages we can observe the classic changes of periportal cholestatic disease with xanthomatous changes, copper deposits, coppers binding protein and sometimes Mallory-Denk bodies.
- Ductopenia is the final consequence of ductal destruction resulting from inflammation and granulomas. It is seen in stages 3 and 4.
- If there is already cirrhosis, it frequently is the biliary type with irregular nodules. Rarely the cirrhotic nodules are quite rounded. They look like puzzle pieces with periseptal peripheral halos, xanthomatous change and copper deposits or of its binding protein.
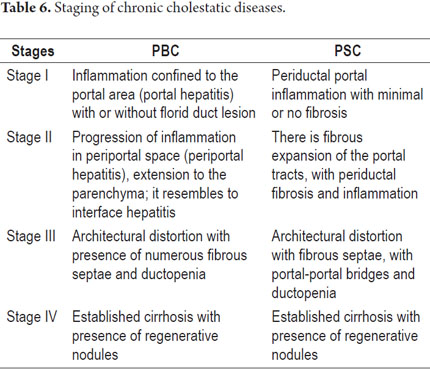
The histological lesions are divided into four stages of PBC (Table 6) (17). The staging is controversial, especially because values can be very heterogeneous and because various stages of the condition can coexist simultaneously. However, for most authors the presence bridging fibrosis has shown itself to be a poor prognostic sign.
Some subtypes of PBC have been described:
- Autoimmune Cholangitis or Negative Primary Biliary Cirrhosis AMA: The clinical, biochemical and pathological profiles are very similar to those of classical PBC, but with negative AMA. This occurs in women who are slightly younger patient. In 10% of cases they are positive for ANA or ASMA.
- Primary Biliary Cirrhosis with ductopenia. The clinical and biochemical profiles from a biopsy shows ductopenia with minimal inflammation and absence of florid duct lesion.
- Primary Biliary Cirrhosis Syndromes overlapping with autoimmune hepatitis in both the clinical and histopathological profiles.
When differential diagnosis of autoimmune hepatitis (AIH) is done can we favor a diagnosis of PBC? Here there are some clues to the diagnosis:
- Both entities have predominant plasmacytic infiltration
- Apoptosis is negligible or absent in PBC
- Interface hepatitis is prominent in AIH
- There is more lymphocytic cholangitis in PBC
- Granulomatous cholangitis is very unusual in AIH
- There is no ductopenia in AIH
- Cirrhosis is biliary type in PBC
PRIMARY SCLEROSING CHOLANGITIS (PSC)
Primary Sclerosing Cholangitis is a chronic cholestatic disease in which there is immune-mediated inflammation and obliteration of segments of the intra and/or extra hepatic biliary tree. These are replaced by fibrous tissue gradually leading to the formation of multiple strictures of the bile duct until terminal liver disease and cirrhosis develop. In 75% of cases both small and large ducts are compromised. In 15% only small ducts are compromised, and in 10% of cases only large ducts are damaged. PSC can occur at any age, but typically occurs after the fourth decade of life. It occurs slightly more often among men than among women. It is closely related to other autoimmune diseases such as chronic inflammatory bowel disease, ulcerative colitis (70%) and Crohn's disease (5-10%). It is diagnosed before the first manifestation of PSC in 75 % of cases, but only 5% of patients with ulcerative colitis have PSC (19, 20).
Diagnosis of PSC has at least three criteria:
1. A cholestatic biochemical profile
2. Magnetic Resonance Cholangiopancreatography (MRCP) shows changes characteristic of multifocal stenosis and segmental dilatation. This is very important. This study has a sensitivity of 80% and specificity of 87%. Endoscopic retrograde cholangiopancreatography (ERCP) can be helpful, but it is not often used or only used in special cases.
3. Exclusion of all probable secondary causes such as bile duct obstruction due to tumors like cholangiocarcinoma, metastasis, stones, IgG4-related cholangitis, recurrent pyogenic cholangitis, infections, ischemic cholangitis, trauma due to preliminary surgery of the bile duct, radiological biliary manipulation. Some of these and the many other causes can lead to terminal stages of Secondary Biliary Cirrhosis (20).
A liver biopsy is usually not necessary for diagnosis and, as in diagnosis of PBC, it has limited uses. It has being used to establish the diagnosis of small duct PSC especially when imaging studies are normal. It has also been used to discard concurrent diseases, to stage lesions, and to monitor progression and response to treatment (21, 22).
What histological changes should we look for?
The initial damage from PSC requires examination of medium and large ducts greater than 100 μ in diameter which cannot be not seen in percutaneous biopsies. Besides looking for segmental lesions, we should look for the following in early stages:
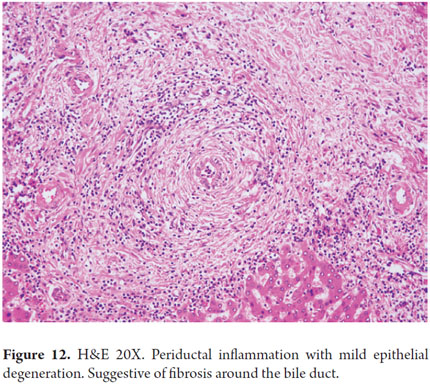
- Pericholangitis and fibrous cholangitis: the presence of mild lymphocytic inflammatory infiltrate with occasional eosinophils in the portal area. There are small numbers of fibrous, edematous stroma surrounding the bile duct (Figures 12 and 13).
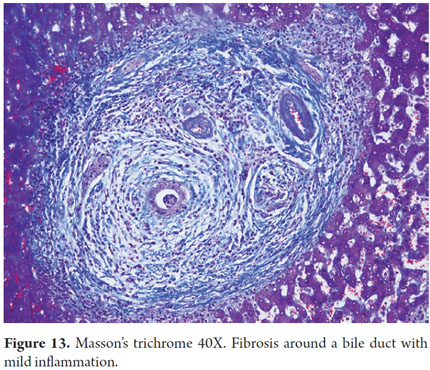
- Lymphocytic infiltrate permeating the ductal epithelium. It is variable in density and segmental.
- Degenerative changes in the ductal epithelium with stratification, cytoplasmic vacuolization, and detachment of the epithelium from the basal membrane.
- Concentric pipes of edematous fibrous tissue surrounding the ducts without obliterating them. This image is known as onion rings (Figure 14).
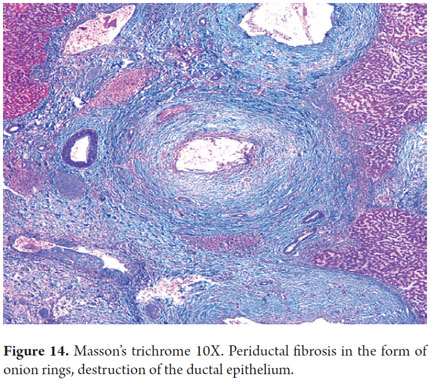
- In the clinical context of chronic cholestasis, when ducts that appear to be normal are found with periductal fibrosis, we must look for lymphocytic cholangitis or duct damage in the serial sections of the biopsy.
In later stages, or as long as the lesion progresses, the following can be observed:
- Periductal fibrosis that progressively surrounds compromised bile ducts and even obliterates them as it becomes sclerotic fibrous tissue which replaces the duct (obliterative fibrosing cholangitis).
- Destruction of the ductal epithelium due to fibrosis with epithelial atrophy or flattening of the epithelium and subsequent ductopenia.
- Cholangiectasias and cholangitic abscesses occur as the result of bile duct damage due to bile deposits. These conditions are very toxic to the epithelium and are usually accompanied by acute inflammation.
- Parenchymal changes associated with cholestasis, bilirubinostasis, ballooning, xanthomatous change, presence of Mallory-Denk bodies, copper deposits and copper binding proteins. Usually there is minimal or no injury to the limiting plate.
- Biliary cirrhosis with periseptal peripheral halos as described for PSC.
Unfortunately, none of these changes is pathognomonic of PSC. There are also segmental changes that require adequate sampling and can occur in other etiologies such as bile duct obstruction from any cause, primary and secondary biliary cirrhosis, bile duct atresia, overlap syndromes, autoimmune hepatitis, graft-versus-host disease, and idiopathic adult ductopenia. Secondary sclerosing cholangitis is the terminal stage of entities such as autoimmune pancreatitis, IgG4-related cholangitis, eosinophilic cholangitis, AIDS cholangiopathy, recurrent pyogenic cholangitis and ischemic cholangitis. Thus, a joint analysis with a complete medical history and diagnostic imaging will always be indispensable.
Several staging systems have been used, none of which have been fully established, but the one described by Ludwig is one of the best accepted (Table 6) (23).
In differential diagnosis of autoimmune hepatitis (AIH) or primary biliary cirrhosis (PBC), should we suspect PSC? The key diagnostic features are:
- Plasmocyte infiltration is much greater in AIH than in PSC, but less than in PBC.
- Necroinflammation, interface hepatitis and apoptosis is negligible or non-existent in the PSC.
- There may be lymphocytic cholangitis in both, but it is greater in PSC.
- Fibrous Obliterative Cholangitis is not observed in AIH.
- Granulomatous cholangitis does not exist in PSC.
- There is no duct loss or ductopenia in AIH.
- Cirrhosis is biliary type in PSC and PBC.
IGG4-RELATED CHOLANGITIS
Systemic disease caused by IgG4 of unknown pathogenesis is characterized by infiltration of plasmocytes that react to IgG4 in immunohistochemical studies, infiltration of T lymphocytes and ductal fibrosis. It compromises numerous organs especially the pancreas (autoimmune pancreatitis) and the bile duct (IgG4 cholangitis). It has high IgG4 serum levels. Its clinical presentation is one of a chronic cholestatic disease, and radiological findings suggest a pancreatic neoplasm. Liver compromise can occur in two ways: mass formation in the hilar region resembling cholangiocarcinoma, and IgG4-related cholangitis resembling primary sclerosing cholangitis producomg stenosis of the intra or extra hepatic bile duct. These entities must be considered in the differential diagnosis. IgG4 cholangitis is not associated with chronic inflammatory bowel disease.
Identification of IgG4-Related Cholangitis is very difficult and requires a high level of suspicion. It responds favorably and rapidly to treatment with steroids (24, 25).
CONCLUSION
Cholestatic processes are definitely a challenge for both the clinician and the pathologist. A biopsy is sometimes an important support for understanding and comprehensive management of patients, but the most important key to the correct diagnosis is adequate clinical pathology.
REFERENCES
1. Li MK, Crawford JM. The pathology of cholestasis. Semin Liver Dis. 2004;24(1):21-42. [ Links ]
2. European Association for the Study of the Liver. EASL Clinical Practice Guidelines: management of cholestatic liver diseases. J Hepatol. 2009;51(2):237-67. [ Links ]
3. Jüngst C, Berg T, Cheng J, Green RM, Jia J, Mason AL, et al. Intrahepatic cholestasis in common chronic liver diseases. Eur J Clin Invest. 2013;43(10):1069-83. [ Links ]
4. Rockey DC, Caldwell SH, Goodman ZD, Nelson RC, Smith AD, American Association for the Study of Liver Diseases. Liver biopsy. Hepatol Baltim Md. 2009;49(3):1017-44. [ Links ]
5. Isse K, Harada K, Nakanuma Y. IL-8 expression by biliary epithelial cells is associated with neutrophilic infiltration and reactive bile ductules. Liver Int Off J Int Assoc Study Liver. 2007;27(5):672-80. [ Links ]
6. Roskams TA, Theise ND, Balabaud C, Bhagat G, Bhathal PS, Bioulac-Sage P, et al. Nomenclature of the finer branches of the biliary tree: canals, ductules, and ductular reactions in human livers. Hepatol Baltim Md. 2004;39(6):1739-45. [ Links ]
7. Lefkowitch JH. Advances in hepatobiliary pathology: update for 2010. Clin Liver Dis. 2010;14(4):747-62. [ Links ]
8. Lefkowitch JH. Hepatobiliary pathology. Curr Opin Gastroenterol. 2008;24(3):269-77. [ Links ]
9. Christoffersen P, Poulsen H. Histological changes in human liver biopsies following extrahepatic biliary obstruction. Acta Pathol Microbiol Scand Suppl. 1970;212:Suppl 212:150+. [ Links ]
10. Pauli-Magnus C, Meier PJ. Hepatobiliary transporters and drug-induced cholestasis. Hepatol Baltim Md. 2006;44(4):778-87. [ Links ]
11. Scheimann AO, Strautnieks SS, Knisely AS, Byrne JA, Thompson RJ, Finegold MJ. Mutations in bile salt export pump (ABCB11) in two children with progressive familial intrahepatic cholestasis and cholangiocarcinoma. J Pediatr. 2007;150(5):556-9. [ Links ]
12. Strautnieks SS, Byrne JA, Pawlikowska L, Cebecauerová D, Rayner A, Dutton L, et al. Severe bile salt export pump deficiency: 82 different ABCB11 mutations in 109 families. Gastroenterology. 2008;134(4):1203-14. [ Links ]
13. López-P R del P, Forero J-D, Sierra F. [Methimazole-induced cholestatic jaundice in a hyperthyroid patient]. Acta Gastroenterol Latinoam. 2014;44(1):52-8. [ Links ]
14. Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1261-73. [ Links ]
15. Flores A, Mayo MJ. Primary biliary cirrhosis in 2014. Curr Opin Gastroenterol. 2014;30(3):245-52. [ Links ]
16. Nguyen DL, Juran BD, Lazaridis KN. Primary biliary cirrhosis. Best Pract Res Clin Gastroenterol. 2010;24(5):647-54. [ Links ]
17. Lindor KD, Gershwin ME, Poupon R, Kaplan M, Bergasa NV, Heathcote EJ, et al. Primary biliary cirrhosis. Hepatol Baltim Md. 2009;50(1):291-308. [ Links ]
18. Jüngst C, Berg T, Cheng J, Green RM, Jia J, Mason AL, et al. Intrahepatic cholestasis in common chronic liver diseases. Eur J Clin Invest. 2013;43(10):1069-83. [ Links ]
19. LaRusso NF, Shneider BL, Black D, Gores GJ, James SP, Doo E, et al. Primary sclerosing cholangitis: summary of a workshop. Hepatol Baltim Md. 2006;44(3):746-64. [ Links ]
20. Chapman R, Fevery J, Kalloo A, Nagorney DM, Boberg KM, Shneider B, et al. Diagnosis and management of primary sclerosing cholangitis. Hepatol Baltim Md. 2010;51(2):660-78. [ Links ]
21. Burak KW, Angulo P, Lindor KD. Is there a role for liver biopsy in primary sclerosing cholangitis? Am J Gastroenterol. 2003;98(5):1155-8. [ Links ]
22. O'Leary JG, Pratt DS. Cholestasis and cholestatic syndromes. Curr Opin Gastroenterol. 2007;23(3):232-6. [ Links ]
23. LaRusso NF, Shneider BL, Black D, Gores GJ, James SP, Doo E, et al. Primary sclerosing cholangitis: summary of a workshop. Hepatol Baltim Md. 2006;44(3):746-64. [ Links ]
24. Webster GJM, Pereira SP, Chapman RW. Autoimmune pancreatitis/IgG4-associated cholangitis and primary sclerosing cholangitis--overlapping or separate diseases? J Hepatol. 2009;51(2):398-402. [ Links ]
25. Kamisawa T, Okamoto A. IgG4-related sclerosing disease. World J Gastroenterol WJG. 2008;14(25):3948-55. [ Links ]
