










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.3 Bogotá Sept. 2014
Presentación de hipertensión portal idiopática en paciente con esclerosis sistémica forma limitada
César Ricardo Ortega Espinosa MD. (1), Lina Maria Saldarriaga Rivera MD. (2)
(1) Internista, Gastroenterólogo. Hospital Universitario Antonio Pedro. Universidad Federal Fluminense. Río de Janeiro, Brasil.
(2) Internista, Reumatóloga. Hospital Clementino Fraga Filho. Universidad Federal de Río de Janeiro, Brasil. E-mail: linamarias7@hotmail.com
Fecha recibido: 06-12-13 Fecha aceptado: 21-07-14
Resumen
La esclerosis sistémica es un trastorno reumático, autoinmune, multisistémico caracterizado por fibrosis de la piel y numerosos órganos. El tracto gastrointestinal es la segunda causa de compromiso sistémico de esta enfermedad, siendo la hipertensión portal idiopática una condición extremadamente rara en estos pacientes. Describimos el caso de una paciente de sexo femenino con alteraciones hepáticas atípicas de hipertensión portal idiopática asociada a esclerosis sistémica forma limitada, entidades que infrecuentemente coexisten en un mismo individuo, permitiéndonos hacer una revisión bibliográfica y reportar esta asociación clínica como la primera en nuestro medio.
Palabras clave
Esclerosis sistémica, hipertensión portal, enfermedad hepática.
INTRODUCCIÓN
La esclerosis sistémica (ES) también denominada esclerodermia es una enfermedad autoinmune del tejido conectivo que afecta más a mujeres entre los 30 y 50 años, caracterizada por inflamación y fibrosis, principalmente cutánea, y puede afectar órganos internos (1).
La ES se clasifica en forma localizada y forma sistémica. La forma localizada involucra lesiones de piel sin compromiso visceral y la forma sistémica se subdivide en forma limitada que incluye el síndrome CREST (calcinosis, Raynaud, esofagopatia, esclerodactilia y telangiectasias), forma difusa y forma visceral sin compromiso cutáneo (2).
El compromiso del tracto gastrointestinal es la segunda causa más frecuente en 80% de los casos. El esófago es la estructura frecuentemente afectada. Los hallazgos comúnmente observados son hipotensión del esfínter esofágico inferior y alteraciones del esófago distal. También han sido identificados gastroparesia, megacolon, pseudoobstrucción y sobrecrecimiento bacteriano (3).
A nivel hepático, la cirrosis biliar primaria es la enfermedad más común, excluyendo drogas hepatotóxicas y hepatitis virales que pueden estar asociadas a la ES forma limitada. La hipertensión portal idiopática es rara en pacientes con enfermedad del tejido conectivo (4).
Describimos el caso de una paciente de sexo femenino, que presentó alteraciones hepáticas atípicas. Lo relevante de este caso es la asociación de la hipertensión portal idiopática con la esclerosis sistémica forma limitada, entidades que infrecuentemente coexisten en un mismo individuo, permitiéndonos hacer una revisión bibliográfica y reportar esta asociación clínica como la primera en nuestro medio.
CASO CLÍNICO
Paciente de sexo femenino, 38 años, blanca, natural de Río de Janeiro, inicia en el 2010 con cuadro clínico de 2 meses de evolución caracterizado por poliartritis inflamatoria de manos, muñecas, codos y rodillas asociada a máculas eritematosas en extremidades, fiebre diaria 38.5 °C, diarrea profusa, debilidad muscular y pérdida de peso involuntaria de 10 kg, sin anorexia y sin sitiofobia.
Al examen físico la paciente presentaba sobrepeso (IMC: 29), rash macular hipercrómico pruriginoso en tórax, dorso y extremidades asociados a engrosamiento moderado de la piel, debilidad muscular proximal simétrica en las cuatro extremidades y hepatomegalia de 3 cm por debajo del margen costal derecho.
Los exámenes de laboratorio reportaron hemograma normal, PCR: 2,64 mg/L, (VR:<3 mg/L), enzimas hepáticas (transaminasas y GGT) normales, C3: 116 mg/dl, C4: 27mg/dl. Factor reumatoide: negativo (Látex y Waaler Rose), anticuerpos antinucleares (ANA): positivo, anticentrómero: positivo y anticuerpos extractables del núcleo (ENAS): negativo (técnica ELISA).
Se realizó serología para citomegalovirus, HIV, toxoplasmosis, clamidia, hepatitis B y C: negativos. La electroforesis de proteínas séricas mostró hipergammaglobulinemia policlonal 28% (2,7g/dl).
Fue realizada radiografía de manos y rodillas sin alteraciones. Biopsia de piel compatible con esclerodermia. Biopsia muscular reportando atrofia muscular de tipo neurogénica, biopsia del nervio sural: neuropatía axonal grave. Electroneuromiografía reporta neuropatía axonal motora de predominio en miembros inferiores. El ecocardiograma evidenció hipertensión pulmonar (PAP: 47mmhg). Examen parasitológico en heces negativo, examen de tránsito de intestino delgado normal. Tomografía abdominal: normal. Gammagrafía esofágica mostró trastornos motores esofágicos moderados. Las pruebas tiroideas, pancreáticas y para enfermedad celiaca (antigliadina, antiendomisio) fueron negativas.
Con estos resultados fue diagnosticado esclerosis sistémica forma limitada e iniciado tratamiento con prednisona 10 mg/día, metotrexato 10mg SC una vez por semana, ácido fólico 5mg/día, bosentán 125 mg dos veces al día y colchicina 1,5 mg/día presentando mejoría importante de la sintomatología clínica.
Después de dos meses de mantener el esquema terapéutico propuesto, la paciente inicia con disnea de pequeños esfuerzos asociada a tos no productiva, telangiectasias en tórax, dorso, extremidades, esclerodactilia y fenómeno de Raynaud.
Fue realizada tomografía de tórax que mostró opacidades en vidrio esmerilado con discreta reticulación en la periferia de los lóbulos inferiores y medios bilateralmente compatible con enfermedad pulmonar intersticial, y se trató con pulsoterapia de ciclofosfamida IV durante 8 meses, con mejora del cuadro clínico.
Posteriormente presentó diarrea severa con hematoquecia, fue indicada colonoscopia que mostró numerosas telangiectasias en todo el recorrido colónico, sin signos de sangrado activo (figura 1).
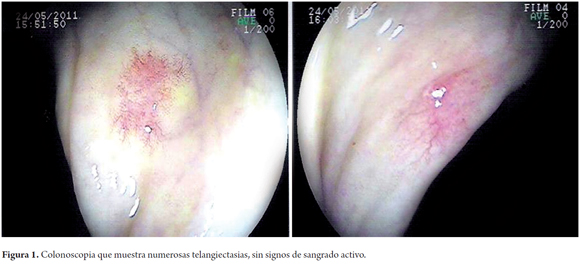
El ecocardiograma no evidenció mejoría en la hipertensión pulmonar (PAP: 46 mmHg). Se encontraron transaminasas discretamente elevadas 1,5 veces el valor normal, bilirrubinas y fosfatasa alcalina normales, anticuerpos para hepatitis autoinmune antimúsculo liso y anti-LKM1 fueron negativos. Anticuerpo antimitocondria positivo 1/80. Niveles de alfa 1 antriptisina y ácido 5 hidroxiindolacético normales.
Debido a la elevación de las enzimas hepáticas, fue suspendido el metotrexato que hasta la fecha tenía una dosis total de 480mg durante 1 año de tratamiento, e indicada biopsia hepática que mostró fibrosis portal con septos delgados, distorsión de la organización lobulillar, células mononucleares ocasionales, algunos puentes capilares necroinflamatorios entre los tractos portales y las venas hepáticas terminales con aumento de volumen de los hepatocitos. La biopsia fue compatible con hipertensión portal idiopática y no demostró toxicidad por metotrexato.
La ecografía abdominal reveló esplenomegalia, la colangiorresonancia no mostró dilatación del tracto biliar y el estudio hemodinámico con cateterismo de suprahepáticas y portografía fue normal, reportando una presión suprahepática enclavada (PSHE) de 5 mmHg, una presión suprahepática libre (PSHL) de 3 mmHg y un gradiente de presión venoso hepático (GPVH) de 2 mmHg.
A pesar de la suspensión del metotrexato, la paciente no presentó mejoría de las transaminasas, evolucionando con hipertensión portal diagnosticada según los criterios propuestos por el Comité de investigación sobre hipertensión portal idiopática de Japón, por la presencia de várices esofágicas de mediano calibre, esplenomegalia y biopsia hepática.
La paciente fue sometida a ligadura endoscópica de las várices esofágicas, por empeoramiento de la hipertensión portal y recibió tratamiento profiláctico con propanolol 40 mg cada 12 h.
DISCUSIÓN
La esclerosis sistémica es un trastorno reumático, autoinmune, multisistémico que afecta principalmente la piel y numerosos órganos. Aunque el compromiso hepático es raro, la enfermedad hepática no se ha considerado un hallazgo significativo de la ES. Varios estudios han demostrado baja prevalencia de patología hepática en poblaciones utilizadas incluso como grupo control. Bartholomew, et al realizaron un estudio con 727 pacientes con ES y encontraron que solo 8 pacientes (1,1%) presentaron compromiso hepático (5).
La hipertensión portal idiopática (HPI) es una condición rara, que suele aparecer a partir de la quinta y sexta década de la vida, con mayor frecuencia en mujeres y suele asociarse a enfermedades autoinmunes. En Occidente se presenta de manera infrecuente y en Oriente se han reportado algunos casos donde la mayor incidencia la tiene Japón con predominio en mujeres, seguido de la India donde ocurre más en hombres entre los 30 y 35 años (6, 7).
La etiología HPI es desconocida. Se han propuesto diferentes teorías que tratan de explicar cómo se desarrolla esta patología, aunque ninguna posee datos suficientemente sólidos; entre ellas se incluyen infecciones crónicas del tracto digestivo que pueden llevar a inflamación portal, así como la exposición a determinados tóxicos como el arsénico, el cloruro de vinilo y fármacos como azatioprina, metotrexate y 6-mercaptopurina. Se ha propuesto también un origen autoinmune debido a mayor incidencia en pacientes con enfermedades del tejido conectivo. Por ejemplo, en la asociación de la ES con la HPI existen diferentes alteraciones inmunológicas en ambas enfermedades, como la disminución de linfocitos supresores o citotóxicos (CD8) y alteración de la relación entre los CD4/CD8 en los pacientes con fibrosis portal no cirrótica (8).
Dilucidar si la asociación es causal o si ambas enfermedades tiene un mismo mecanismo etiopatogénico no es posible en la actualidad. Recientemente se ha descrito la asociación de HPI con la enfermedad celiaca e incluso una disminución de la hipertensión portal tras instaurar una dieta sin gluten (9).
Los criterios propuestos por el Comité de investigación sobre hipertensión portal idiopática de Japón para el diagnóstico de esta entidad incluyen hipertensión portal con esplenomegalia, anemia, generalmente secundaria a hiperesplenismo, evidencia de várices esofágicas por endoscopia o radiología, pruebas de función hepáticas normales o cerca de la normalidad y biopsia sugestiva. Se excluyen cirrosis hepática, trastornos hematológicos, parasitosis hepatobiliar, oclusión de la vena porta y las venas suprahepáticas (10).
El diagnóstico de HPI en nuestra paciente se determinó por la presencia de várices esofágicas demostradas en la endoscopia, esplenomegalia, además de la biopsia. Aunque la paciente no presentó anemia, se evidenció durante la evolución pruebas de función hepática casi normales, lo que coincide con el estudio reportado por Okudaira y colaboradores donde muestra que las pruebas de función hepática pueden estar cerca de parámetros de normalidad, como criterio de inclusión diagnóstico de HPI (11).
En cuanto a la biopsia, existen algunas características que permiten el diagnóstico de HPI. Ludwig, et al realizaron un estudio con 25 muestras de biopsia hepáticas con HPI, y encontraron dilatación de las venas portales con herniación de estas en el parénquima hepático circundante, arquitectura acinar alterada, puentes capilares y necroinflamatorios entre los tractos portales y las venas hepáticas terminales y en menor frecuencia fueron identificados formación de megasinusoides con distribución aleatoria, desplazamiento y anomalía de las ramas de la vena hepática con o sin flebosclerosis y septos fibrosos delgados (12).
La biopsia de nuestra paciente demostraba la existencia de fibrosis portal con septos delgados, distorsión de la organización lobulillar y algunos puentes capilares necroinflamatorios entre los tractos portales y las venas hepáticas hallazgos compatibles con lo demostrado por Ludwing y colaboradores para HPI. No se observaron nódulos que sugirieran la existencia de hiperplasia nodular regenerativa. Tampoco se demostró toxicidad por metotrexato.
Como criterios de exclusión fueron descartados cirrosis biliar primaria, parasitosis y enfermedades hematológicas. La ecografía abdominal con Doppler descartó obstrucción hepática de la vena porta y la colangiorresonancia no mostró dilatación del tracto biliar.
Los pacientes con hipertensión portal sinusoidal tienen un gradiente de presión venosa hepática (GPVH) > de 5 mmHg, mientras que si el origen es presinusoidal el GPVH suele ser normal (trombosis portal, hipertensión portal idiopática, estadios iniciales de la cirrosis biliar primaria y esquistosomiasis). En nuestro caso, la paciente presentó un estudio de hemodinamia hepática normal con un GPVH de 2 mmHg, debido al estadio inicial de la enfermedad, lo que coincide con lo reportado por Wongcharatrawee, et al que explican que en la etapa inicial de la HPI, el estudio hemodinámico con cateterismo de suprahepáticas y portografía puede ser normal (13).
Se ha reconocido que algunos fármacos hepatotóxicos como azatioprina y metotrexato son inductores de fibrosis hepática llevando a hipertensión portal. En nuestro caso no se utilizaron dosis altas de metotrexato siendo suspendido en el momento en que se elevaron las enzimas hepáticas.
La dosis tóxica acumulada del metotrexato se ha estimado en 3 gramos (14). Nuestra paciente recibió solo una sexta parte de esa dosis (480 mg) durante un año de tratamiento y posterior a la retirada del fármaco mostró signos de empeoramiento de hipertensión portal, lo cual nos lleva a deducir que la dosis de metotrexato dada a la paciente no fue el agente etiológico de la hipertensión portal, debido a que fueron utilizadas dosis bajas, por tiempo no prolongado y con reporte de biopsia sin hepatotoxicidad.
Las manifestaciones hepáticas en las enfermedades reumáticas autoinmunes son poco estudiados y la prevalencia es variable. El pronóstico en estos pacientes es más favorable que en aquellos que padecen de hipertensión portal secundaria a cirrosis. El diagnóstico precoz permite instaurar un tratamiento oportuno, modificando la evolución, incrementando la supervivencia a 90% y reduciendo la mortalidad generada por el sangrado de vías digestivas.
La paciente fue diagnosticada por reumatología con esclerosis sistémica forma limitada, presentando síndrome CREST con fenómeno de Raynaud, esofagopatía, esclerodactilia, telangiectasias cutáneas en tórax, dorso, extremidades y telangiectasias colónicas sin signos de sangrado activo, y complicaciones derivadas de su patología de base como la hipertensión pulmonar, enfermedad pulmonar intersticial y trastorno en la motilidad esofágica, las cuales mejoraron con el tratamiento. Además de la hipertensión portal idiopática tratada en este caso.
Luego de una búsqueda en diferentes base de datos, entre ellas Pubmed, Cochrane y Lilacs (palabras clave hipertensión portal idiopática, esclerosis sistémica y esclerodermia) con fecha de corte 27 de noviembre de 2013, se encontraron solo 7 artículos que describen la asociación de la hipertensión portal idiopática con la esclerosis sistémica, la mayoría reportados en Japón (15-21). Siendo estas entidades excepcionales coexistentes en un mismo individuo, no existen casos reportados en nuestro medio, lo que sería la primera descripción clínica realizada en Latinoamérica.
Declaración de fuentes de financiación y posible conflicto de intereses
Financiamiento: No se recibió patrocinio de ningún tipo para llevar a cabo este artículo.
Conflicto de intereses: Los autores declaran no tener ningún conflicto de intereses.
REFERENCIAS
1. Black CM. Scleroderma-clinical aspects. J Intern Med 1993; 234(2): 115-18. [ Links ]
2. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988; 15: 202-5. [ Links ]
3. Domsyc R, Fasanella K, Bielefeldt K. Gastrointestinal Manifestations of Systemic Sclerosis, Dig Dis and Sciences 2008; 53(5): 1163-74. [ Links ]
4. Buffet C, Husson JM. Liver and generalized scleroderma. Nouv Presse Med 1974; 28(3): 2701-2. [ Links ]
5. Bartholomew LG, Cain JC, Winkelmann RK, Baggenstoss AH. Chronic disease of the liver associated with systemic scleroderma. Am J Dig Dis 1964; 9: 43-55. [ Links ]
6. Harmanci O, Bayraktar Y. Clinical characteristics of idiophatic portal hypertension. World J Gastroenterol 2007; 13(13): 1906-11. [ Links ]
7. Vassia M, Curciarello JO, Corrons F, Viola, M, Zamboni E, Castagno M, et al. Hipertensión portal idiopática con infarto esplénico. Complicación no descripta. Acta Gastroenterol Latinoam 2001; 31(1): 27-30 [ Links ]
8. Nayyar AK, Sharma BK, Sarin SK, Malhotra P, Broor SL, Sachdev G. Characterization of peripheral blood lymphocytes in patients with non-cirrhotic portal fibrosis: a comparison with cirrhotics and healthy controls. J Gastroenterol Hepatol 1990; 5(5): 554-9. [ Links ]
9. Zamami F, Amiri A, Shakeri R, Zare A, Mohamadnejad M. Celiac disease as a potential cause of idiopathic portal hypertension: a case report. Journal of Medical Case Reports 2009; 3:68-71. [ Links ]
10. Okuda K, Nakashima T, Okudaira M, Kage M, Aida Y, Omata M. Anatomic basis of hepatic venographic alterations in idiopathic portal hypertension. Liver 1981; 1: 255-63. [ Links ]
11. Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Sem Liver Dis 2002; 22: 59-71. [ Links ]
12. Ludwing J, Hashimoto E, Obata H, Baldus WP. Idiopathic portal hypertension: a histopathological study of 26 Japanese cases. Histopathol 1993; 22: 227-234. [ Links ]
13. Wongcharatrawee S, Groszmann R. Hemodynamic assessment in clinical practice in portal hypertensive cirrhotics. Annals of gastroenterology 2001, 14(3): 158-65. [ Links ]
14. Montaudié H, Sbidian E, Paul C, Maza A, Gallini A, Aractingi S, et al. Methotrexate in psoriasis: a systematic review of treatment modalities, incidence, risk factors and monitoring of liver toxicity. J Eur Acad Dermatol Venereol 2011; 25(2): 12-8. [ Links ]
15. Umeyama K, Yui S, Fukamizu A, Yoshikawa K, Yamashita T. Idiopathic hypertension associated with progresive systemic sclerosis. Am J Gastroenteral 1982; 77: 645-8. [ Links ]
16. Sánchez Manuel J, Seco Gil JL, Sáez-Royuela F, Llanos Chavarri MC, Pérez álvarez JL, García Plata E. Idiopathic portal hypertension, multiple focal nodular hepatic hyperplasia, the CREST syndrome and protein S deficiency. Rev Esp Enferm Dig 1997; 89: 55-9. [ Links ]
17. Watanabe Y, Mizukami T, Egawa T, Okamoto S, Sakauchi M, Takita T, et al. A case of progressive systemic sclerosis complicated by idiopathic portal hypertension with severe anemia. Ryumachi 1999; 39: 586-90. [ Links ]
18. John Moschos, Grigoris I Leontiadis, Clive Kelly, James Henry, Savvas Kadis. Idiopathic portal hypertension complicating systemic sclerosis: a case report. BMC Gastroenterology. 2005; 5(1): 16-19. [ Links ]
19. Takagi K, Nishio S, Akimoto K, Yoshino T, Kawai S. A case of systemic sclerosis complicated by idiopathic portal hypertension: case report and literature review. Mod Rheumatol. 2006; 16(3): 183-7. [ Links ]
20. Kogawa H, Migita K, Ito M, Takii Y, Daikoku M, Nakao M, et al. Idiopathic portal hypertension associated with systemic sclerosis and Sjögrens syndrome. Clin Rheumatol 2005; 24(5): 544-7. [ Links ]
21. Pérez García C, Coll S, Blanch Rubió J. Hipertensión portal idiopática asociada a esclerosis sistémica progresiva. Rev Esp Reumatol 2002; 29(2): 65-68. [ Links ]
