










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.3 Bogotá Sept. 2014
Presentation of Idiopathic Portal Hypertension in a Patient with Limited Systemic Sclerosis
César Ricardo Ortega Espinosa MD. (1), Lina Maria Saldarriaga Rivera MD. (2)
(1) Internist and Gastroenterologist at Hospital Universitario Antonio Pedro and Universidad Federal Fluminense in Rio de Janeiro, Brazil.
(2) Internist and Rheumatologist at Hospital Clementino Fraga Filho and the Universidad Federal de Rio de Janeiro in Rio de Janeiro, Brazil. E-mail: linamarias7@hotmail.com
Received: 06-12-13 Accepted: 21-07-14
Abstract
Systemic sclerosis is a multisystem rheumatic autoimmune disorder characterized by fibrosis of the skin and many other organs. The gastrointestinal tract is the second cause of systemic compromise of this disease, but idiopathic portal hypertension is an extremely rare condition in these patients. We describe the case of a female patient with atypical hepatic changes of idiopathic portal hypertension associated with limited systemic sclerosis. Since these entities rarely coexist in the same individual, and since this is the first clinical association in our environment, we present this literature review and case report.
Keywords
Systemic sclerosis, portal hypertension, liver disease.
INTRODUCTION
Systemic sclerosis (SS) is also called scleroderma. It is an autoimmune disease of the connective tissue that mostly affects women between 30 and 50 years of age. It is primarily characterized by inflammation and dermal fibrosis and can affect internal organs (1).
SS is classified into localized and systemic forms. The localized form involves skin lesions without visceral involvement. The systemic form is divided into a limited form called the CREST syndrome (calcinosis, Raynaud phenomenon, esophagopathy, sclerodactyly and telangiectasia), a diffuse form and a visceral form without cutaneous involvement (2).
Compromise of the gastrointestinal tract is the second most common cause in 80% of cases. The affected structure is frequently the esophagus. Findings commonly observed are hypotension of the lower esophageal sphincter and distal esophagus alterations. Gastroparesis, megacolon, pseudo-obstruction and bacterial overgrowth have also been identified (3).
In the liver, primary biliary cirrhosis is the most common illness, excluding viral hepatitis and hepatotoxic drugs that may be associated with SS in a limited way. Idiopathic portal hypertension is rare in patients with connective tissue disease (4).
We describe the case of a female patient who presented atypical hepatic impairment. The relevance of this case is the association of idiopathic portal hypertension with limited systemic sclerosis. These entities rarely coexist in the same individual, and this is the first report of this clinical association in our country. We present this case together with a review of the literature.
CASE REPORT
The patient was a 38 year old white woman who was born in Rio de Janeiro. In 2010 she came to the hospital after two months clinical evolution characterized by inflammatory polyarthritis of her hands, wrists, elbows and knees associated with erythematous macules on her extremities, a daily fever of 38.5 C, profuse diarrhea, muscular weakness and involuntary weight loss of 10 kg. She had no signs of anorexia or sitophobia.
A physical examination showed that the patient was overweight (BMI: 29), presented a pruritic hyperchromic maculopapular rash on her chest, back and limbs which was associated with moderate skin thickening, had symmetrical proximal muscular weakness in all four extremities, and had 3 cm hepatomegaly below the right costal margin.
Laboratory tests reported normal blood counts: PCR: 2.64 mg/L, RV <3mg/L, normal liver enzymes (transaminases and GGT), C3: 116mg/dl, C4: 27 mg/dl, rheumatoid factor: negative (latex test and Waaler-Rose test), antinuclear antibodies (ANA): positive, anti-centromere: positive, and extractable nuclear antibodies (ENAS): negative (ELISA technique).
Blood tests for cytomegalovirus, HIV, toxoplasmosis, chlamydia, hepatitis B and hepatitis C were all negative. Serum protein electrophoresis showed 28% polyclonal hypergammaglobulinemia (2.7 g/dl).
Radiography of the patients hands and knees showed no alterations. A skin biopsy was compatible with scleroderma. A muscle biopsy showed neurogenic type muscular atrophy. A sural nerve biopsy showed severe axonal neuropathy. Electroneuromyography showed axonal motor neuropathy predominantly in the lower limbs. Echocardiography showed pulmonary hypertension (PAP: 47 mmHg). Stool tests for parasites were negative, and an examination of small intestine transit was normal. An abdominal examination was normal. Esophageal scintigraphy showed moderate esophageal motor disorders. Thyroid, pancreatic and celiac disease (anti-gliadin, anti-endomysium) tests were all negative.
Based on these results, limited systemic sclerosis was diagnosed. Treatment was initiated with 10 mg/day of prednisone, 10 mg/week of subcutaneous methotrexate, 5 mg/day of folic acid, 125mg of bosentan twice daily and 1.5 mg/day of colchicine. The patient began to show significant improvement of her clinical symptoms.
After two months of this therapeutic regimen, upon slight exertion the patient began to experience dyspnea which was associated with a nonproductive cough, sclerodactyly, Raynauds phenomenon and telangiectasia of the chest, back and extremities.
A chest CT scan showed ground glass opacities with discrete crosslinking in the periphery of the lower and medium lobes which were bilaterally compatible with interstitial lung disease. This was treated with cyclophosphamide IV pulse therapy for eight months which resulted in improvement of symptoms.
Later the patient presented severe diarrhea with hematochezia. Colonoscopy showed numerous telangiectasias around the colonic route but without active bleeding (Figure 1).
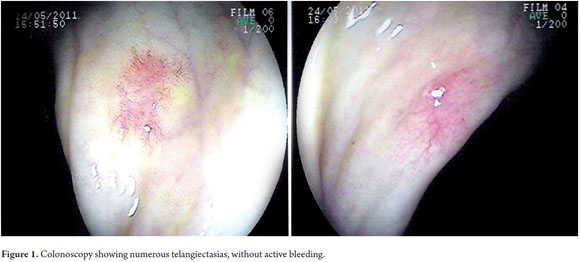
The echocardiogram showed no improvement in pulmonary hypertension (PAP 46 mmHg). Transaminases were slightly elevated at 1.5 times the normal value; alkaline phosphatase bilirubin was normal; antibodies to autoimmune hepatitis, anti-smooth muscle and anti-LKM1 were negative; anti-mitochondrial antibody was positive at 1/80. Alpha-1 antitrypsin and 5 hydroxyindoleacetic acid levels were found.
Due to elevated liver enzymes, methotrexate was suspended. To this point the patient had received a total dose of 480 mg over 1 year of treatment. A liver biopsy showed portal fibrosis with thin septa, distortion of lobular organization, occasional mononuclear cells, necroinflammatory capillary bridges between portal tracts and terminal hepatic veins with increased volume of hepatocytes. The biopsy was consistent with idiopathic portal hypertension but showed no methotrexate toxicity.
Abdominal ultrasound revealed splenomegaly, and magnetic resonance cholangiopancreatography (MRCP) showed no dilatation of the biliary tract, A hemodynamic study with catheterization of the hepatic vein and portography was normal. It reported hepatic venous wedge pressure (HVWP)) of 5 mmHg, free hepatic venous pressure of 3 mmHg and a hepatic venous pressure gradient (HVPG) of 2 mmHg.
Despite discontinuation of methotrexate, the patients transaminase levels did not improve and evolved until portal hypertension was diagnosed according to the criteria proposed by the Research Committee on Idiopathic Portal Hypertension of Japan (presence of esophageal varices of medium caliber, splenomegaly and liver biopsy).
The patient underwent endoscopic ligation of esophageal varices due to worsening of portal hypertension and received prophylactic treatment with 40mg of propranolol every 12 hours.
DISCUSSION
Systemic sclerosis is a rheumatic, autoimmune, multisystem disorder that primarily affects the skin and many other organs. Because hepatic involvement is rare, liver disease has not been considered a significant finding related to SS. Several studies have shown low prevalences of liver disease in these populations, and they have even been used as control groups. Bartholomew et al. conducted a study involving 727 patients with SS and found that only 8 patients (1.1%) had liver involvement (5).
Idiopathic portal hypertension (IPH) is a rare condition that usually appears during the fifth and sixth decades of life, most often in women. It is often associated with autoimmune diseases. In the West, it occurs infrequently while the East has reported some cases with the highest incidence in Japan where it is most common in women. The second highest incidence is in India where it occurs most often in men between 30 and 35 years of age (6, 7).
The etiology of IPH is unknown. Different theories that attempt to explain how this disease develops have been proposed, but none of them have enough solid data about chronic digestive tract infections that can lead to portal inflammation or about exposure to certain toxic substances such as arsenic, vinyl chloride and drugs such as azathioprine, methotrexate and 6-mercaptopurine. An autoimmune basis has also been proposed on the basis of the higher incidence in patients with connective tissue diseases. For example, there are various immunological abnormalities shared by SS and IPH such as decreased suppressor and cytotoxic lymphocytes (CD8) and an altered CD4/CD8 ratio in patients with non-cirrhotic portal fibrosis (8).
At present, it is not possible to determine whether the association between SS and IPH is causal, or whether both diseases have the same etiopathogenetic mechanism. Recently, an association of celiac disease with IPH has been described including decreased portal hypertension after establishing a gluten free diet (9).
The criteria proposed by the Research Committee on Idiopathic Portal Hypertension of Japan for the diagnosis of this entity include portal hypertension with splenomegaly, anemia, usually secondary to hypersplenism, endoscopic or radiological evidence of esophageal varices, normal or near normal liver function tests and suggestive biopsy. Liver cirrhosis, hematological disorders, hepatobiliary parasitosis, occlusion of the portal vein and hepatic veins are excluded (10).
Diagnosis of IPH in our patient was through endoscopic identification of esophageal varices, the presence of splenomegaly, and the results of a biopsy. Although the patient did not present anemia, idiopathic portal hypertension was evidenced during liver function tests even though the test results were nearly normal. This is consistent with a by Okudaira et al. which shows that liver function tests close to normal parameters may be used as an inclusion criterion in the diagnosis of IPH (11).
Some features of the biopsy allow a diagnosis of IPH. Ludwig et al. conducted a study of 25 liver biopsy samples from patients with IPH and found dilated portal veins which were herniated in the surrounding hepatic parenchyma. They also found altered acinar architecture and necroinflammatory capillary bridges between portal tracts and terminal hepatic veins. Less frequently, they found randomly distributed megasinusoids, thin fibrous septa, and displacement or failure of branches of the hepatic vein with or without phlebosclerosis (12).
The biopsy of our patient demonstrated the existence of portal fibrosis with thin septa, distortion of lobular organization, necroinflammation and some capillary bridges between portal tracts and hepatic veins. All of this was consistent with the findings of Ludwig and colleagues regarding IPH. No observations were made that suggested the existence of nodular regenerative hyperplasia or that demonstrated toxicity due to methotrexate.
Primary biliary cirrhosis, hematological diseases and parasites were discarded as exclusion criteria. Abdominal Doppler ultrasound ruled out obstruction of the hepatic portal vein, and cholangiography showed no dilatation of the biliary tract.
Patients with sinusoidal portal hypertension have a hepatic venous pressure gradient (HVPG)> 5mmHg, whereas presinusoidal HVPG is usually normal (portal thrombosis, idiopathic portal hypertension, early stages of primary biliary cirrhosis and schistosomiasis). In our case, the patient had a normal hepatic hemodynamic study with an HVPG of 2 mmHg. This was because this patient was in the initial stage of the disease which is consistent with the explanation of Wongcharatrawee et al. who explained that the hemodynamic study using hepatic catheterization and portography may be normal in the initial stage of IPH (13).
Some hepatotoxic drugs such as azathioprine and methotrexate are recognized as inducers of hepatic fibrosis leading to portal hypertension. In our case, high doses of methotrexate were not used, and the drug was suspended when liver enzymes became elevated.
The cumulative toxic dose of methotrexate is estimated to be 3 grams (14). Our patient received only 480mg, one sixth of that dose, over the course of a year of treatment. After drug withdrawal, she showed signs of worsening portal hypertension which led us to conclude that methotrexate was not the etiological agent of her portal hypertension since low doses had been used over a long period of time and because of the biopsy report which showed no hepatotoxicity.
Hepatic manifestations of autoimmune rheumatic diseases are poorly studied, and their prevalence varies. The prognoses for these patients are more favorable than those for patients with portal hypertension secondary to cirrhosis. Early establishment of diagnosis allows prompt treatment, modification of the conditions evolution, an increase of the survival rate to 90%, and a reduction of mortality caused by bleeding in the gastrointestinal tract.
Our patient had been diagnosed by the Rheumatology Department with limited systemic sclerosis and CREST syndrome with Raynauds phenomenon; esophagopathy; sclerodactyly; cutaneous telangiectasias in the chest, back, limbs and colonic telangiectasias without active bleeding. Besides the idiopathic portal hypertension discusses in this article, our patient presented complications due to underlying diseases including pulmonary hypertension, interstitial lung disease and esophageal motility disorder. All of these improved with treatment.
A search of databases including PubMed, Cochrane, and Lilacs using the keywords, idiopathic portal hypertension, systemic sclerosis and scleroderma was conducted on November 27, 2013. Just seven articles, mostly from Japan, describing the association of idiopathic portal hypertension with systemic sclerosis were found (15-21). Although these exceptional entities can coexist in one individual, there had been no cases reported in our country. This is the first clinical description in Latin America.
Declaration of sources of funding and potential conflicts of interest
Funding: No funding of any kind was received for this article.
Conflicts of interest: The authors declare no conflicts of interest.
REFERENCES
1. Black CM. Scleroderma-clinical aspects. J Intern Med 1993; 234(2): 115-18. [ Links ]
2. LeRoy EC, Black C, Fleischmajer R, Jablonska S, Krieg T, Medsger TA Jr, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988; 15: 202-5. [ Links ]
3. Domsyc R, Fasanella K, Bielefeldt K. Gastrointestinal Manifestations of Systemic Sclerosis, Dig Dis and Sciences 2008; 53(5): 1163-74. [ Links ]
4. Buffet C, Husson JM. Liver and generalized scleroderma. Nouv Presse Med 1974; 28(3): 2701-2. [ Links ]
5. Bartholomew LG, Cain JC, Winkelmann RK, Baggenstoss AH. Chronic disease of the liver associated with systemic scleroderma. Am J Dig Dis 1964; 9: 43-55. [ Links ]
6. Harmanci O, Bayraktar Y. Clinical characteristics of idiophatic portal hypertension. World J Gastroenterol 2007; 13(13): 1906-11. [ Links ]
7. Vassia M, Curciarello JO, Corrons F, Viola, M, Zamboni E, Castagno M, et al. Hipertensión portal idiopática con infarto esplénico. Complicación no descripta. Acta Gastroenterol Latinoam 2001; 31(1): 27-30 [ Links ]
8. Nayyar AK, Sharma BK, Sarin SK, Malhotra P, Broor SL, Sachdev G. Characterization of peripheral blood lymphocytes in patients with non-cirrhotic portal fibrosis: a comparison with cirrhotics and healthy controls. J Gastroenterol Hepatol 1990; 5(5): 554-9. [ Links ]
9. Zamami F, Amiri A, Shakeri R, Zare A, Mohamadnejad M. Celiac disease as a potential cause of idiopathic portal hypertension: a case report. Journal of Medical Case Reports 2009; 3:68-71. [ Links ]
10. Okuda K, Nakashima T, Okudaira M, Kage M, Aida Y, Omata M. Anatomic basis of hepatic venographic alterations in idiopathic portal hypertension. Liver 1981; 1: 255-63. [ Links ]
11. Okudaira M, Ohbu M, Okuda K. Idiopathic portal hypertension and its pathology. Sem Liver Dis 2002; 22: 59-71. [ Links ]
12. Ludwing J, Hashimoto E, Obata H, Baldus WP. Idiopathic portal hypertension: a histopathological study of 26 Japanese cases. Histopathol 1993; 22: 227-234. [ Links ]
13. Wongcharatrawee S, Groszmann R. Hemodynamic assessment in clinical practice in portal hypertensive cirrhotics. Annals of gastroenterology 2001, 14(3): 158-65. [ Links ]
14. Montaudié H, Sbidian E, Paul C, Maza A, Gallini A, Aractingi S, et al. Methotrexate in psoriasis: a systematic review of treatment modalities, incidence, risk factors and monitoring of liver toxicity. J Eur Acad Dermatol Venereol 2011; 25(2): 12-8. [ Links ]
15. Umeyama K, Yui S, Fukamizu A, Yoshikawa K, Yamashita T. Idiopathic hypertension associated with progresive systemic sclerosis. Am J Gastroenteral 1982; 77: 645-8. [ Links ]
16. Sánchez Manuel J, Seco Gil JL, Sáez-Royuela F, Llanos Chavarri MC, Pérez álvarez JL, García Plata E. Idiopathic portal hypertension, multiple focal nodular hepatic hyperplasia, the CREST syndrome and protein S deficiency. Rev Esp Enferm Dig 1997; 89: 55-9. [ Links ]
17. Watanabe Y, Mizukami T, Egawa T, Okamoto S, Sakauchi M, Takita T, et al. A case of progressive systemic sclerosis complicated by idiopathic portal hypertension with severe anemia. Ryumachi 1999; 39: 586-90. [ Links ]
18. John Moschos, Grigoris I Leontiadis, Clive Kelly, James Henry, Savvas Kadis. Idiopathic portal hypertension complicating systemic sclerosis: a case report. BMC Gastroenterology. 2005; 5(1): 16-19. [ Links ]
19. Takagi K, Nishio S, Akimoto K, Yoshino T, Kawai S. A case of systemic sclerosis complicated by idiopathic portal hypertension: case report and literature review. Mod Rheumatol. 2006; 16(3): 183-7. [ Links ]
20. Kogawa H, Migita K, Ito M, Takii Y, Daikoku M, Nakao M, et al. Idiopathic portal hypertension associated with systemic sclerosis and Sjögrens syndrome. Clin Rheumatol 2005; 24(5): 544-7. [ Links ]
21. Pérez García C, Coll S, Blanch Rubió J. Hipertensión portal idiopática asociada a esclerosis sistémica progresiva. Rev Esp Reumatol 2002; 29(2): 65-68. [ Links ]
