










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.3 Bogotá Sept. 2014
Colestasis neonatal e infantil. Aproximación al diagnóstico histopatológico
Rocío del Pilar López Panqueva MD. (1), Lina Eugenia Jaramillo Barberi MD. (2)
(1) Patóloga Hospital Universitario Fundación Santa Fe de Bogotá. Universidad de Los Andes. Bogotá, Colombia.
(2) Patóloga Hospital de La Misericordia. Universidad Nacional de Colombia. Bogotá, Colombia.
Fecha recibido: 07-08-14 Fecha aceptado: 19-08-14
Resumen
Aun cuando el papel de la biopsia hepática está cambiando con el desarrollo de nuevos métodos de diagnóstico y del avance de las técnicas de imagen, de biomarcadores no invasivos, estudios proteómicos y genómicos, la biopsia hepática realizada en el momento y con la indicación adecuadas continúa siendo una importante herramienta para la evaluación y diagnóstico de los niños con colestasis tanto en el periodo neonatal como durante la infancia temprana o tardía, no solo para determinar una etiología o establecer un pronóstico sino para guiar una terapia (1).
Son múltiples las causas y varios los patrones morfológicos observados, puede estar relacionada a un defecto genético del metabolismo hepático incluyendo la síntesis de ácidos biliares, la formación y función de transportadores de membrana o a alteraciones en el desarrollo de las vías biliares, muchos de los cuales pueden sobreponerse y deben interpretarse en conjunto con los hallazgos clínicos, genéticos y de laboratorio. Los síndromes heredados que producen colestasis intrahepática y la atresia biliar son las causas más comunes de enfermedad hepática crónica y la indicación principal para el trasplante hepático en niños.
El enfoque que aquí daremos hace hincapié en la estrecha colaboración que debe existir entre pediatras, gastroenterólogos, cirujanos pediátricos y los patólogos para la correcta identificación y posterior manejo sea médico o quirúrgico incluyendo el trasplante hepático, de muchas de las patologías colestásicas que afectan este grupo etario (2, 3).
Palabras clave
Colestasis, biopsia hepática, atresia de la vía biliar, hepatitis neonatal, deficiencia de alfa-1- antitripsina, ductopenia.
INTRODUCCIÓN
En los niños, las enfermedades que se manifiestan con colestasis a menudo son el resultado de procesos patológicos que inician en la vida intrauterina o postnatal temprana, cuando aún el hígado no ha alcanzado una completa madurez funcional; esto favorece la susceptibilidad a agresiones tanto exógenas como endógenas. Además, estudios recientes nos dan las bases moleculares de fenotipos clínicos, identificando genes candidatos o mutaciones genéticas asociadas, que modifican la enfermedad y explican muchas de las entidades causantes de colestasis en la infancia.
COLESTASIS NEONATAL
La ictericia neonatal que se prolonga más de 14 días después del nacimiento es inusual. La incidencia de colestasis neonatal es de aproximadamente 1 de cada 2.500 nacidos vivos y por lo tanto es baja la frecuencia de casos que pueden ser vistos por la mayoría de los proveedores de atención médica primaria de los niños (4).
Es fundamental establecer si la ictericia del recién nacido es causada por condiciones no colestásicas o si se trata de una ictericia colestásica definida como la presencia de hiperbilirrubinemia conjugada (más de 2,0 mg/dl o más de 20% de la cifra de bilirrubina total). La ictericia aparece en los primeros 90 días especialmente en las primeras 2 semanas de vida, existiendo muchísimas causas tanto intrahepáticas como extrahepáticas. Establecer una correcta etiología es considerada una urgencia, ya que está asociada con un alto riesgo de muerte o de complicaciones que requiere un diagnóstico temprano, con el fin de iniciar un tratamiento que puede ser salvador de vida (5).
El diagnóstico diferencial de la colestasis neonatal es muy amplio y requiere un enfoque gradual basado en la historia clínica y el examen físico, estudios de laboratorio e imagenológicos para identificar rápidamente la etiología subyacente. El reconocimiento temprano de la colestasis neonatal es esencial para asegurar el tratamiento oportuno y mejorar el pronóstico, así como para determinar la necesidad de realizar estudios complementarios especializados al paciente y/o su familia (2).
En la tabla 1 enumeramos solo algunas de ellas y nos referiremos en este artículo a las más comunes: atresia de la vía biliar extrahepática, hepatitis neonatal idiopática, la deficiencia de alfa 1 antitripsina y la ductopenia.

¿Es posible diferenciar atresia de la vía biliar y hepatitis neonatal idiopática?
No existe un test específico que nos dé la clave diagnóstica entre estas dos entidades que constituyen casi 70-80% de todas las colestasis de la infancia, entidades que requieren un manejo muy diferente; por lo cual la historia clínica, estudios de imágenes diagnósticas y en ocasiones la biopsia hepática interpretada por un patólogo experimentado, puede aportar información muy útil y establecer en más de 90% de los casos un diagnóstico acertado (6).
ATRESIA DE LA VÍA BILIAR (AB)
La causa más frecuente de colestasis neonatal es la atresia de la vía biliar extrahepática (AB), constituyendo cerca de la tercera parte de los casos de colestasis neonatal, con una incidencia de 1:15.000 nacidos vivos. Es un proceso idiopático, inflamatorio y destructivo de los ductos biliares extra e intrahepáticos que lleva a fibrosis y obstrucción ductal progresiva y evoluciona hasta cirrosis de tipo biliar. En esta condición el tratamiento quirúrgico oportuno, procedimiento de Kasai o portoenterostomía, realizado antes de los primeros 100 días de vida puede definir el curso de la enfermedad cuya historia natural es variable, siendo impredecible la velocidad de la progresión o el pronóstico de esta entidad considerada la primera causa (50% de los casos) de trasplante hepático pediátrico y fatal si no es tratada (1, 4, 7-9).
Su patogénesis no está clara, se describen dos formas diferentes:
1. La forma fetal /embrionaria, la más grave de ellas, en la que se sugiere que es secundaria a un evento adverso durante la embriogénesis y se caracteriza por la aparición de colestasis desde el nacimiento y la ausencia de remanente de vía biliar. Corresponde a 10-35% de los casos y hasta en 20% de estos se acompaña de otras malformaciones como poliesplenia, defectos cardiovasculares, situs inversus, malrotación intestinal.
2. Forma postnatal o perinatal, en la cual se postula una obliteración ductal adquirida, no acompañada de malformaciones, observándose remanentes de la vía biliar en la porta hepatitis y con desarrollo más tardío de cirrosis. Se han postulado sin poder corroborarlo, el papel de algunas infecciones virales como reovirus 3, rotavirus y citomegalovirus, o formas genéticas dado el aumento del riesgo encontrado en algunas familias (3, 10).
3. Ninguna de las características histopatológicas observadas es patognomónica de la atresia biliar, por lo cual es absolutamente indispensable la correlación con los datos clínicos y de laboratorio, estudios de imágenes preoperatorios y colangiografía intraoperatoria que ayuden a confirmar los cambios obliterativos ductales y la ausencia de vesícula biliar.
Los cambios morfológicos más observados en la atresia de la vía biliar son:
- Proliferación ductular con presencia de ductos irregulares en su forma, anastomosados, dando una apariencia que semeja una cadena (figuras 1A, B y C) este hallazgo es el más descrito, en la forma fetal/embrionaria y se considera un indicador de pobre pronóstico (11).
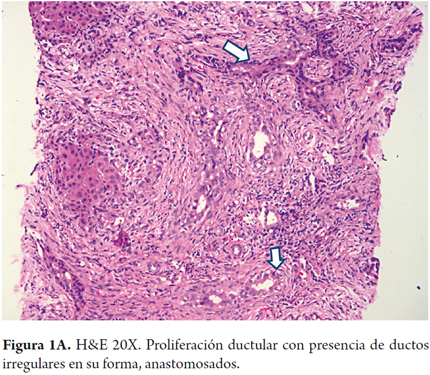
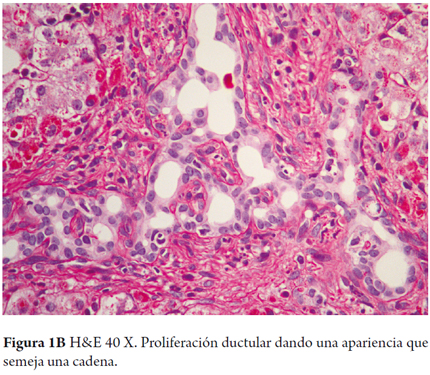
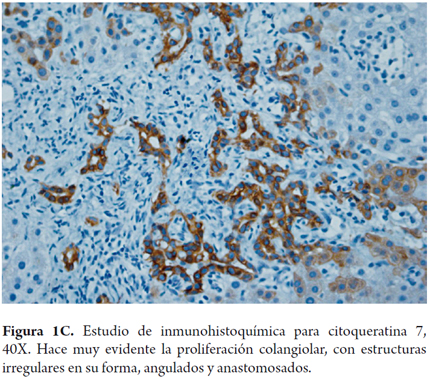
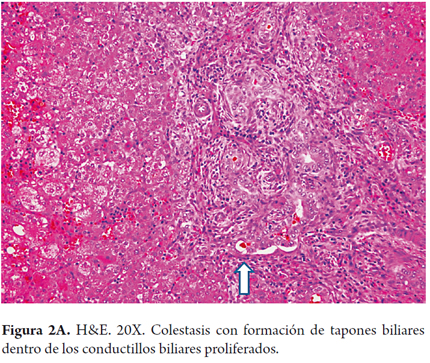
- Colestasis intracanalicular con formación de tapones biliares dentro de los conductillos biliares (figura 2A) e intracanaliculares (figura 2B).
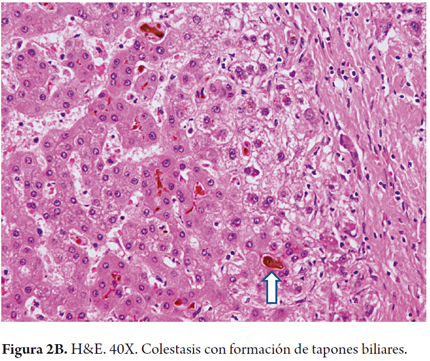
- Hematopoyesis extramedular, está relacionada con estrés durante el periodo intrauterino y neonatal.
- Edema y fibrosis portal.
- Cirrosis de tipo biliar en estadios avanzados (figura 3).
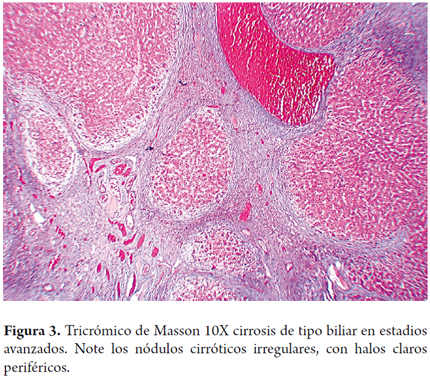
Cuando la biopsia se lleva a cabo antes de las 6 semanas de edad estas características pueden no estar presentes y la colangiografía intraoperatoria con nuevas biopsias pueden ser requeridas para confirmar el diagnóstico (5).
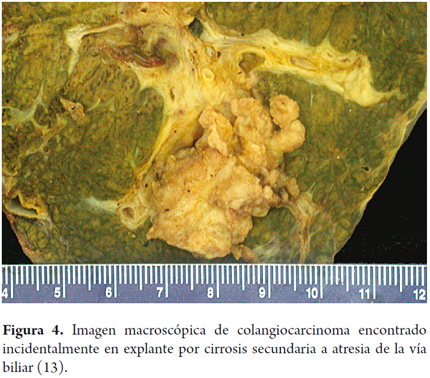
Todos los pacientes desarrollan fibrosis progresiva hasta la cirrosis, 35% de los cuales sobreviven en promedio de 10 años, posterior al procedimiento de Kasai y antes de necesitar trasplante hepático. Una de las principales complicaciones de los pacientes con AB y Kasai es la colangitis recurrente, además de un aumento del riesgo de hepatocarcinoma, rara vez hepatoblastoma o colangiocarcinoma (figura 4) (12, 13).
HEPATITIS NEONATAL, HEPATITIS NEONATAL GIGANTOCELULAR O HEPATITIS NEONATAL IDIOPáTICA (HNI)
Ante la imposibilidad de demostrar una etiología específica, en aproximadamente 35-40% de los casos de colestasis prolongada neonatal, el diagnóstico de HNI debe ser considerado; es entonces un diagnóstico de exclusión. Entre 15-20% son casos familiares, los restantes ocurren en forma esporádica y la tercera parte de ellos cursa como una hepatitis fulminante y se han descrito casos de hepatocarcinoma en fases tardías (14). En la tabla 2 se resume el comportamiento HNI.

Los niños presentan ictericia progresiva y persistente de patrón colestásico, coluria, acolia o hipocolia y hepatomegalia. Según el grado de alteración de la síntesis hepática presentará coagulopatía hasta falla hepática fulminante (15).
Muchas posibles etiologías de esta patología, desde agentes infecciosos que incluyen todas las consideradas infecciones congénitas o microorganismos STORCH, paramyxovirus, HIV, papilomavirus (PVH); también este patrón es visto en la hepatitis asociada a anemia hemolítica, en la nutrición parenteral total y en algunas enfermedades metabólicas (16, 17).
Es difícil establecer un diagnóstico teniendo en cuenta solo la morfología, no hay cambios patognomónicos, algunas de las características histopatológicas son compartidas con otras entidades causantes de colestasis en especial con la AB, las más observadas son:
- Compromiso panlobulillar con transformación gigantocelular en la que los hepatocitos son muy grandes con numerosos núcleos, (entre 3 y 20 núcleos); representa una respuesta hepática a la lesión especialmente visto en este grupo etario (figuras 5A y B). Es inusual en adultos.
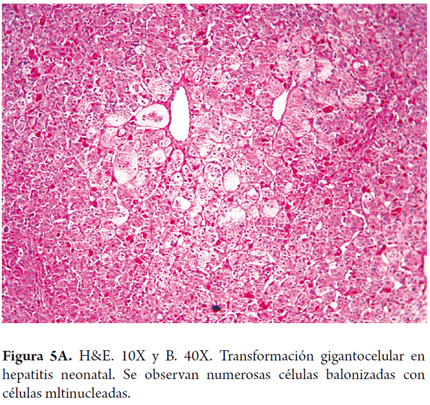
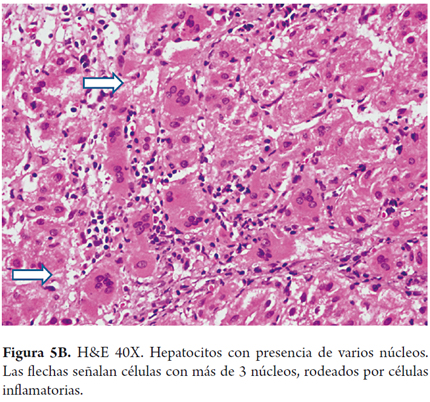
- Proliferación ductal o colangiolar.
- Presencia de depósitos de bilis en conductillos biliares formando tapones.
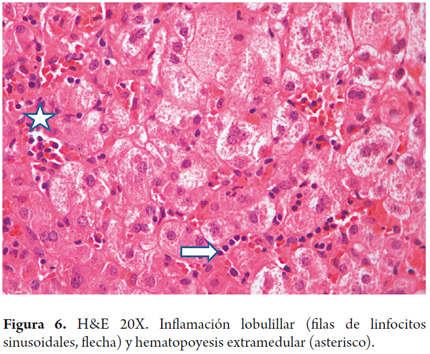
Inflamación lobulillar con linfocitos y neutrófilos también con hematopoyesis extramedular (figura 6).
- Depósitos de hierro intrahepatocitarios.
- Balonización hepatocelular con transformación pseudoacinar (figura 7).
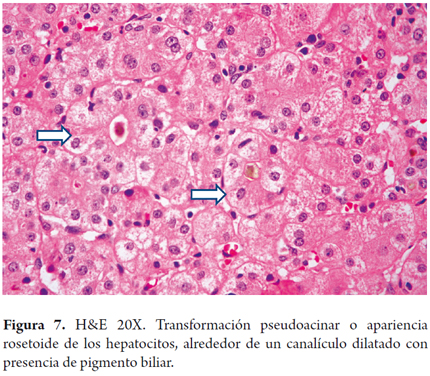
- Fibrosis inicialmente portal con posterior formación de puentes.
DEFICIENCIA DE ALFA 1 ANTITRIPSINA (Α1AT)
Esta entidad es no solamente la más frecuente causa genética de enfermedad hepática en los niños, sino que además tiene una importante predisposición al desarrollo de enfermedad hepática crónica y hepatocarcinoma en la vida adulta. Su incidencia es de 1 por cada 2.000-5.000 nacidos vivos y tiene patrón de herencia autosómica codominante (recesiva) y no ligada al sexo. Es causada por una mutación en la proteína α1ATZ que se encuentra en el retículo endoplásmico de la célula hepatocitaria y es secretada a la circulación en forma monomérica. El gen está localizado en el cromosoma 14 en la región q31-32,3, conocido como gen Serpina1 o PI con más de 100 variantes la gran mayoría sin ningún significado clínico, llamadas variantes genéticas normales o fenotipo PiM, de sus 6 subtipos (M1 al M6) el M1 es encontrado entre 80-90% de la población, hay variantes alélicas deficientes PiS (concentración α1AT <60%), PiZ (concentración α1AT <15%) encontradas especialmente en Europa y USA, otras variantes son disfuncionales; todas ellas pueden producir enfermedad respiratoria y/o hepática (18, 19).
La enfermedad clínica se manifiesta más frecuentemente en pacientes homocigotos nulos, ZZ o SS y menos en los heterocigotos MZ o SZ, a su vez con variantes rápidas (α1AT-F), medias (α1AT- M), lentas (α1AT-S)o muy lentas (α1AT-Z) determinado según sea la velocidad de movilidad electroforética de la α1AT. La tabla 3 resume los diferentes fenotipos de la α1AT (20).

Las variantes más asociadas a enfermedad hepática son los homocigotos PiZZ de los cuales hasta 15% tienen enfermedad antes de los 20 años, PiM, PiS y los heterocigotos PiSZ, PiMZ. El espectro de la enfermedad hepática es muy amplio manifestándose en el periodo neonatal con colestasis y un cuadro de hepatitis neonatal, siendo una de las principales causantes de trasplante hepático en este grupo etario, posteriormente se presenta como hepatitis aguda o crónica, colestasis con disminución de ductos biliares intrahepáticos y cirrosis con o sin hepatocarcinoma.
El gen mutante Z dirige la síntesis de grandes cantidades de la proteína Z mutante en el hígado y se acumula intracelularmente, en lugar de ser secretada a la circulación, este acúmulo dentro de los hepatocitos causa la lesión hepática a través de una cascada de la apoptosis hepatocelular, regeneración y muerte (21).
¿Cuándo debemos sospechar la presencia de deficiencia de α1AT si solo 30% de los pacientes homocigotos progresan a falla hepática o cirrosis? La respuesta es en todo niño con colestasis neonatal, cuando hay cirrosis en la infancia o juvenil o cuando las pruebas de función hepática estén alteradas persistentemente (22-25).
Los hallazgos histopatológicos son usualmente subestimados, el diagnóstico correcto se basa en una adecuada correlación clínico-patológica, biopsia y estudios de laboratorio. En las primeras 12 semanas de la vida las alteraciones morfológicas son sutiles, en más de 80% son semejantes a los de una HNI, por lo que la biopsia por sí sola es insuficiente para establecer el diagnóstico.
- Se basa en encontrar: la presencia de glóbulos eosinofílicos demostrados con estudios de histoquímica y/o inmunohistoquímica que representan las glicoproteínas acumuladas en el retículo endoplásmico; son PAS positivos resistentes a la digestión con diastasa y están localizados en el citoplasma de los hepatocitos de la zona periportal (figuras 8).
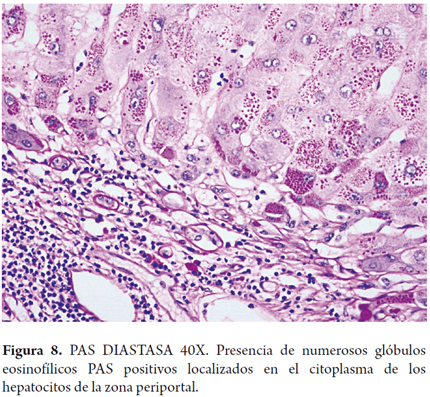
- Los restantes hallazgos lo comparten con otras etiologías como la inflamación portal y fibrosis que inicia en los espacios porta hasta la formación de nódulos de regeneración y cirrosis.
En bebés menores de 12-13 semanas los glóbulos de α1AT pueden ser muy escasos e incluso no estar presentes por lo cual el diagnóstico debe ir encaminado a la determinación de los niveles séricos de α1AT y a establecer su fenotipo. La exactitud diagnóstica de los glóbulos Pas-D positivos es para los fenotipos PiZ, es cercana a 99% en niños con enfermedad grave y a 60% cuando la enfermedad tiene moderada severidad, mientras que en los restantes puede no encontrarse (1, 26).
DUCTOPENIA
Esta entidad incluye un grupo heterogéneo de enfermedades caracterizadas por la reducción en el número de los ductos biliares intrahepáticos pequeños; recibe otros nombres como hipoplasia y escasez de ductos biliares intrahepáticos.
Tiene una condición bien definida conocida como síndrome de Alagille o displasia arteriohepática que se asocia a anomalías cardíacas, faciales, oculares y esqueléticas pero puede ocurrir igualmente como una entidad no sindromática que se encuentra frecuentemente relacionada con otras condiciones entre las que se incluyen infecciones especialmente por rubéola y CMV, enfermedades metabólicas como el déficit de Alfa -1 antitripsina y alteraciones cromosómicas como el síndrome de Down.
La patogénesis en incierta y aunque se considera una entidad que destruye progresivamente los ductos biliares, su evolución es variable pudiendo resolver espontáneamente, producir fibrosis e incluso desarrollar cirrosis.
El diagnóstico se basa en encontrar diminución marcada o ausencia del número de ductos biliares interlobulares (figura 9); para establecer el diagnóstico la relación ducto biliar, espacio porta debe ser de 0,4 o menos (27, 28).
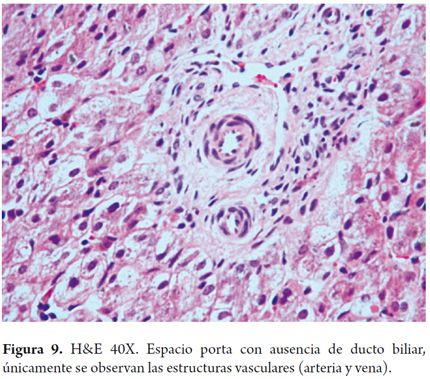
Otros hallazgos histopatológicos son compartidos con otras entidades causantes de colestasis y son:
- Colestasis canalicular (figura 10)
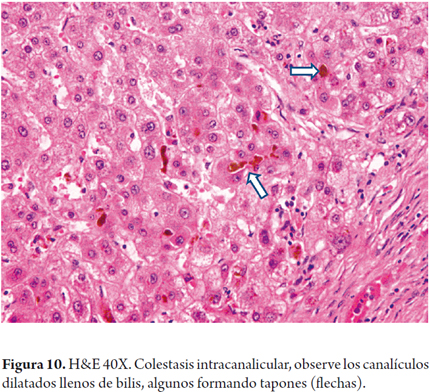
- Espacios porta pequeños o inaparentes
- Balonización hepatocelular con ocasional transformación gigantocelular
- Focos de hematopoyesis extramedular
- Fibrosis inicialmente portal y periportal de grado variable que puede evolucionar a cirrosis.
CONCLUSIÓN
Las enfermedades colestásicas del recién nacido y de la infancia son difíciles de diagnosticar tanto desde el punto de vista clínico como desde el histopatológico. Requieren el análisis conjunto de una exhaustiva historia clínica, conocer los antecedentes familiares, estudios de imágenes, laboratorios usualmente especializados, estudios de biología molecular, genéticos y, en algunos casos, necesitan de una biopsia hepática para poder realizar una adecuada correlación clínico-patológica.
REFERENCIAS
1. Ovchinsky N, Moreira RK, Lefkowitch JH, Lavine JE. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012; 19(4): 250-62. [ Links ]
2. Feldman AG, Sokol RJ. Neonatal Cholestasis. Neoreviews 2013; 14(2). [ Links ]
3. Santos JL, Choquette M, Bezerra JA. Cholestatic liver disease in children. Curr Gastroenterol Rep 2010; 12(1): 30-9. [ Links ]
4. McKiernan PJ. Neonatal cholestasis. Semin Neonatol 2002; 7(2): 153-65. [ Links ]
5. Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2004; 39(2): 115-28. [ Links ]
6. Dehghani SM, Haghighat M, Imanieh MH, Geramizadeh B. Comparison of different diagnostic methods in infants with Cholestasis. World J Gastroenterol 2006; 12(36): 5893-6. [ Links ]
7. Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr 2003; 37(1): 4-21. [ Links ]
8. Shinkai M, Ohhama Y, Take H, Kitagawa N, Kudo H, Mochizuki K, et al. Long-term outcome of children with biliary atresia who were not transplanted after the Kasai operation: >20-year experience at a childrens hospital. J Pediatr Gastroenterol Nutr 2009; 48(4): 443-50. [ Links ]
9. Pereira TN, Walsh MJ, Lewindon PJ, Ramm GA. Paediatric cholestatic liver disease: Diagnosis, assessment of disease progression and mechanisms of fibrogenesis. World J Gastrointest Pathophysiol. 2010; 1(2): 69-84. [ Links ]
10. Venigalla S, Gourley GR. Neonatal cholestasis. Semin Perinatol 2004; 28(5): 348-55. [ Links ]
11. Low Y, Vijayan V, Tan CE. The prognostic value of ductal plate malformation and other histologic parameters in biliary atresia: an immunohistochemical study. J Pediatr 2001; 139(2): 320-2. [ Links ]
12. Tatekawa Y, Asonuma K, Uemoto S, Inomata Y, Tanaka K. Liver transplantation for biliary atresia associated with malignant hepatic tumors. J Pediatr Surg 2001; 36(3): 436-9. [ Links ]
13. Vera A, Villaveces D, López R. Orthotopic liver transplantation for biliary atresia complicated by incidental cholangiocarcinoma. J Pediatr Gastroenterol Nutr 2012; 55(3): 336-7. [ Links ]
14. Moore L, Bourne AJ, Moore DJ, Preston H, Byard RW. Hepatocellular carcinoma following neonatal hepatitis. Pediatr Pathol Lab Med 1997; 17(4): 601-10. [ Links ]
15. Correa KK, Nanjundiah P, Wirtschafter DD, Alshak NS. Idiopathic neonatal giant cell hepatitis presenting with acute hepatic failure on postnatal day one. J Perinatol 2002; 22(3): 249-51. [ Links ]
16. Drut R, Gómez MA, Drut RM, Lojo MM. Human papillomavirus (HPV)-associated neonatal giant cell hepatitis (NGCH). Pediatr Pathol Lab Med 1996; 16(3): 403-12. [ Links ]
17. Domiati-Saad R, Dawson DB, Margraf LR, Finegold MJ, Weinberg AG, Rogers BB. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol 2000; 3(4): 367-73. [ Links ]
18. Kalsheker NA. alpha1-Antitrypsin deficiency: best clinical practice. J Clin Pathol 2009; 62(10): 865-9. [ Links ]
19. Zarrilli F, Elce A, Scorza M, Giordano S, Amato F, Castaldo G. An update on laboratory diagnosis of liver inherited diseases. Biomed Res Int 2013. [ Links ]
20. Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet 2005; 365(9478): 2225-36. [ Links ]
21. Teckman JH, Jain A. Advances in alpha-1-antitrypsin deficiency liver disease. Curr Gastroenterol Rep 2014; 16(1): 367. [ Links ]
22. Chappell S, Hadzic N, Stockley R, Guetta-Baranes T, Morgan K, Kalsheker N. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology 2008; 47(1): 127-32. [ Links ]
23. Perlmutter DH, Brodsky JL, Balistreri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review. Hepatology 2007; 45(5): 1313-23. [ Links ]
24. Campbell KM, Arya G, Ryckman FC, Alonso M, Tiao G, Balistreri WF, et al. High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J Pediatr Gastroenterol Nutr 2007; 44(1): 99-103. [ Links ]
25. De Serres FJ. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ Health Perspect 2003; 111(16): 1851-4. [ Links ]
26. Topic A, Prokic D, Stankovic I. Alpha-1-antitrypsin deficiency in early childhood. Fetal Pediatr Pathol 2011; 30(5): 312-9. [ Links ]
27. Alagille D. Estrada A, Hadchouel et al. Syndrome paucity of interlobular bile ducts Alagille syndrome of arteriohepatic dysplasia): Review of 80 cases. The Journal of Pediatrics 1987; 110 (2): 195-200. [ Links ]
28. De Tommaso AM, Kawasaki AS, Hessel G. Paucity of intrahepatic bile ducts in infancy. Experience of a tertiary center. Arq Gastroenterol 2004; 41(3): 190-192. [ Links ]
