










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.29 no.3 Bogotá Sept. 2014
Neonatal and Infantile Cholestasis: An Approach to Histopathological Diagnosis
Rocío del Pilar López Panqueva MD. (1), Lina Eugenia Jaramillo Barberi MD. (2)
(1) Pathologist at the Hospital Universitario Fundación Santa Fe de Bogotá and the Universidad de Los Andes in Bogotá, Colombia.
(2) Pathologist at the Hospital de La Misericordia and the Universidad Nacional de Colombia in Bogotá, Colombia.
Received: 07-08-14 Accepted: 19-08-14
Abstract
Although the role of liver biopsies is changing with the development of new diagnostic methods and advances in imaging techniques, non-invasive biomarkers, proteomic and genomic studies, a liver biopsy performed at the right time and with appropriate indications continues to be an important tool for assessment and diagnosis of children with cholestasis. This is equally true in the neonatal period, in early childhood, and in late childhood not only for determination of an etiology and establishing a prognosis, but also for guiding treatment of the patient (1).
There are multiple causes and morphological patterns that may be related to a genetic defect in aspects of hepatic metabolism including synthesis of bile acids, formation and function of membrane transporters, and alterations in the development of the bile ducts. Many of these may overlap and should be interpreted in conjunction with clinical, genetic and laboratory findings. Inherited syndromes that produce intrahepatic cholestasis and biliary atresia are the most common causes of chronic liver disease and the leading indication for liver transplantation in children.
The approach we present here emphasizes the close cooperation that should exist between pediatricians, gastroenterologists, pediatric surgeons and pathologists for proper identification of many of the cholestatic diseases that can affect this age group. Subsequent surgical or medical management may include liver transplantation (2, 3).
Keywords
Cholestasis, liver biopsy, biliary atresia, neonatal hepatitis, Alpha-1 antitrypsin, ductopenia.
INTRODUCTION
In children, diseases manifested with cholestasis are often the result of pathological processes that begin in utero or in early postnatal life when the liver has not yet reached full functional maturity. This favors susceptibility to both exogenous and endogenous aggressions. Recent studies have provided the molecular basis of clinical phenotypes and have identified candidate genes which modify disease and associated genetic mutations which can explain many of the entities causing cholestasis in infancy.
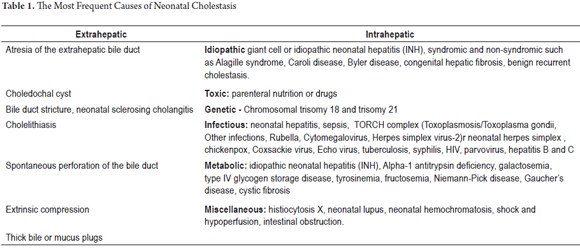
NEONATAL CHOLESTASIS
Neonatal jaundice that lasts more than 14 days after birth is unusual. The incidence of neonatal cholestasis is approximately 1 in 2,500 live births, so most providers of primary health care for children rarely see these cases (4).
It is essential to establish whether newborn jaundice is non-cholestatic or cholestatic h (defined as the presence of more than 2.0 mg/dl conjugated hyperbilirubinemia, or more than 20% of total bilirubin). Jaundice occurs in the first 90 days of life, especially in the first 2 weeks. There are many causes, both intrahepatic and extrahepatic. Establishing the correct etiology is urgent as hyperbilirubinemia is associated with a high risk of death or complications, so early diagnosis is necessary in order to initiate treatment that may save a life (5).
The differential diagnosis of neonatal cholestasis is very broad and requires a gradual approach based on clinical history, physical examination, laboratory tests and imaging to quickly identify the underlying etiology. Early recognition of neonatal cholestasis is essential to ensure timely treatment, improve the prognosis and determine the need for specialized complementary studies of the patient and/or family (2).
In Table 1 we list only some of them, and in this article we will refer to the most common: atresia of the extrahepatic bile duct, idiopathic neonatal hepatitis, alpha-1 antitrypsin deficiency and ductopenia.
Is it possible to differentiate biliary atresia from idiopathic neonatal hepatitis?
These two entities constitute 70% to 80% of all childhood cholestasis but they require very different kinds of management. Since there is no specific test that is the key to differentiating between them, the clinical history, imaging studies and sometimes s liver biopsy performed by an experienced pathologist must can provide the information used. In more than 90% of cases an accurate diagnosis is established (6).
BILIARY ATRESIA
The most common cause of neonatal cholestasis is atresia of the extrahepatic bile duct which accounts for nearly a third of all cases of neonatal cholestasis. Its incidence is 1:15,000 live births. It is an idiopathic, inflammatory and destructive process in the extrahepatic and intrahepatic bile ducts which leads to progressive fibrosis and ductal obstruction and then to biliary type cirrhosis. In this condition the appropriate surgical treatment is performance of a hepatoportoenterostomy (also known as a Kasai procedure) within the first 100 days of life before the course of the disease is set. This diseases natural history varies unpredictably, but it is the reason for 50% of all pediatric liver transplants and can be fatal if untreated (1, 4, 7-9).
Its pathogenesis is unclear, but two different forms have been described.
1. The fetal/embryonic form is the most severe. It has been suggested that it is secondary to an adverse event during embryogenesis. It is characterized by cholestasis at birth and the absence of remnant bile ducts. Fetal cholestasis accounts for 10% to 35% of all cases, and up to 20% of these are accompanied by other malformations such as polysplenia, cardiovascular defects, situs inversus, and intestinal malrotation.
2. In postnatal or perinatal cholestasis it is postulated that an acquired ductal obliteration occurs unaccompanied by malformations. In these cases bile duct remnants are observed in the porta hepatis and there is no later development of cirrhosis. It has been postulated that viral infections such as reovirus type 3, rotavirus and cytomegalovirus or genetic factors may play roles in this type of cholestasis, but these hypotheses have not been confirmed. However, there is increased risk of this condition in some families (3, 10).
3. None of the histopathological features are pathognomonic of biliary atresia, so correlation with clinical and laboratory data, preoperative imaging studies and intraoperative cholangiography is absolutely essential to help confirm the ductal obliteration and the absence of the gallbladder.
The most frequently observed morphological changes in biliary atresia are:
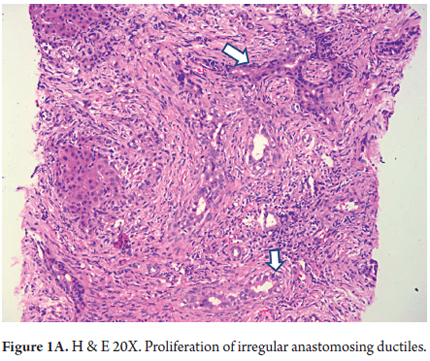
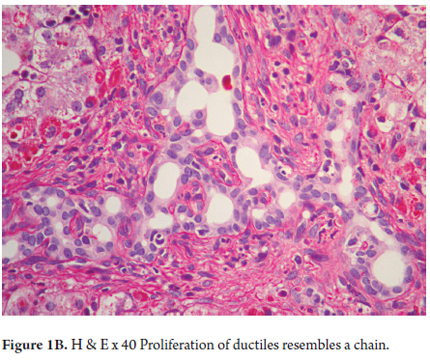
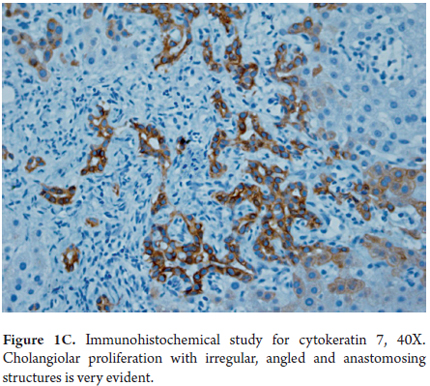
- Proliferation of ducts, and the presence of abnormal ducts with anastomoses that has the appearance of a chain (Figures 1 A, B and C). This has been most often described in the fetal/embryonic form and is considered to be an indicator of poor prognosis. (11)
- Intracanalicular cholestasis with bile plugs within bile ducts and canaliculi (Figures 2 A and B)
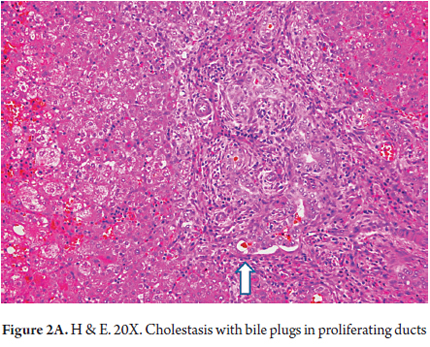
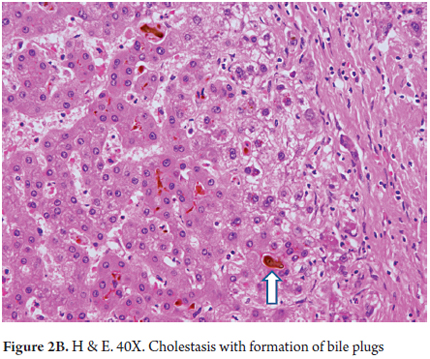
- Extramedullary hematopoiesis which is related to stress during the intrauterine and neonatal period.
- Edema and portal fibrosis
- Advanced stages of biliary type cirrhosis (Figure 3)
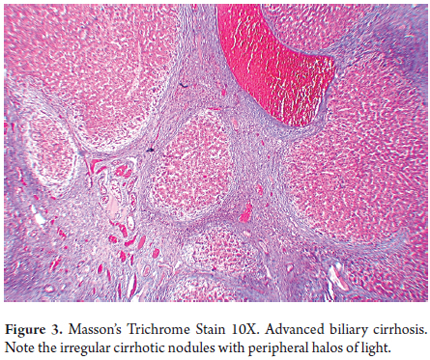
When a biopsy is taken before 6 weeks of age these features may not be present, so that intraoperative cholangiography with new biopsies may be required to confirm the diagnosis (5).
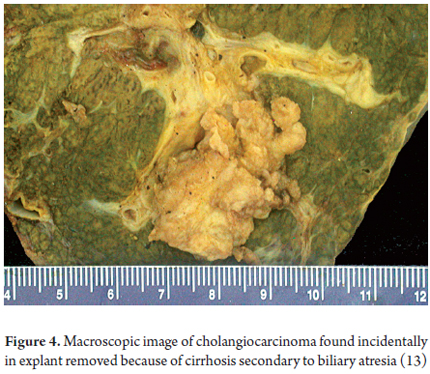
All patients develop fibrosis which progresses until it becomes cirrhosis. 35% of patients survive an average of 10 years after a hepatoportoenterostomy and before they need liver transplantation. One of the major complications in patients with biliary atresia who undergo a hepatoportoenterostomy is recurrent cholangitis. In addition they have high a risk of hepatocellular carcinoma and in rare cases even develop cholangiocarcinoma or hepatoblastoma (Figure 4) (12, 13).
NEONATAL HEPATITIS, NEONATAL GIANT CELL HEPATITIS AND IDIOPATHIC NEONATAL HEPATITIS (INH)
A diagnosis of idiopathic neonatal hepatitis (INH) should be considered when a specific etiology cannot be demonstrated (as happens in approximately 35% to 40% of all cases of prolonged neonatal cholestasis). At this point diagnosis is made by exclusion. 15% to 20% are familial cases. The remaining cases occur sporadically, but one third are develop as fulminant hepatitis, and cases of hepatocellular carcinoma have been reported in late stages (14). Table 2 summarizes the behavior of INH.

Children with INH have progressive jaundice and a persistent cholestatic pattern with choluria, acholia, decreased bile secretion and hepatomegaly. Depending on the degree of impairment of hepatic synthesis, coagulopathy will appear until fulminant hepatic failure occurs (15).
Many possible etiologies have been considered for this disease including infectious agents considered to be congenital infections such as TORCH microorganisms, paramyxoviruses, HIV and papillomavirus (PVH). This pattern is also seen in hepatitis associated with hemolytic anemia, in total parenteral nutrition, and in some metabolic diseases (16, 17).
It is difficult to establish a diagnosis solely on the basis of morphology, and there are no pathognomonic changes, and some of the histopathological features of INH are shared with other entities causing cholestasis especially with biliary atresia. The most common are:
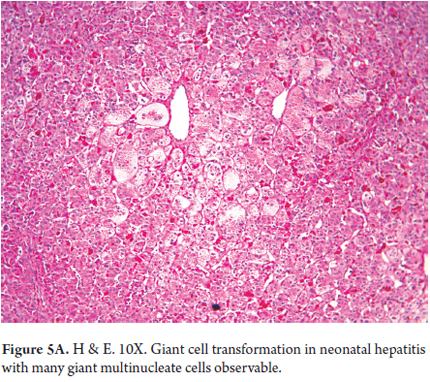
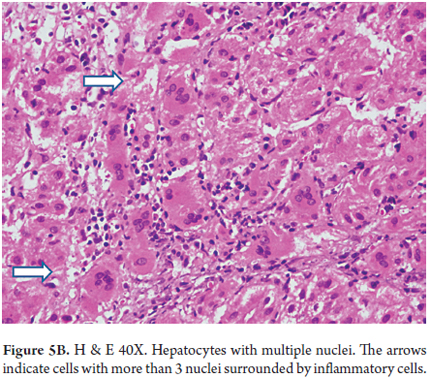
- Panlobular compromise with giant cell transformation in which hepatocytes are very large with many nuclei (between 3 and 20 nuclei). This represents a hepatic response to damage and is seen especially in this age group (Figures 5 A and B). It is unusual in adults.
- Ductal or cholangiolar proliferation
- Presence of bile deposits forming plugs in bile ducts
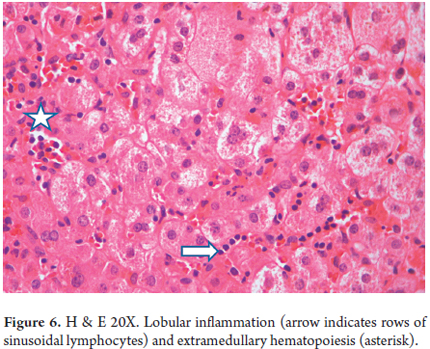
- Lobular inflammation with lymphocytes, neutrophils, and extramedullary hematopoiesis (Figure 6)
- Iron deposits within hepatocytes
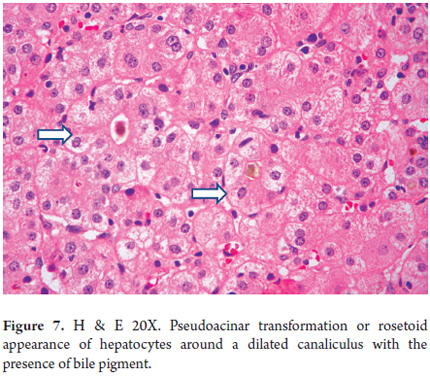
- Hepatocellular ballooning with pseudo-acinar transformation (Figure 7)
- Portal fibrosis leading to subsequent formation of bridges
ALPHA 1 ANTITRYPSIN (Α1AT) DEFICIENCY
This entity is not only the most common genetic cause of liver disease in children, but also has significantly predisposes the development of chronic liver disease and hepatocellular carcinoma in adulthood. Its incidence is 1 in 2,000 to 5,000 live births. It has an autosomal codominant pattern (recessive) and is not sex-linked. Alpha 1 antitrypsin deficiency is caused by changes in the α1ATZ protein found in the endoplasmic reticulum of hepatocytes. α1AT is secreted into the blood circulation as a monomer. Mutations of the SERPINA1 (serpin peptidase inhibitor, clade A (alpha-1 antiproteinase, antitrypsin), member 1) gene are gene responsible for these changes. The cytogenetic location of SERPINA1 is 14q32.1 (molecular location on chromosome 14: base pairs 94,376,746 to 94,390,691). Although it has more than 100 variants, the majority have no clinical significance and are called normal gene variants or PiM phenotype. Of its 6 subtypes (M1 to M6) M1 is found in between 80% 90% of the population. There are PiS variants in which α1AT concentrations are less than 60% and PiZ variants in which α1AT concentrations are less than 15%. These are found especially in Europe and the USA. Other variants are dysfunctional and can all cause respiratory and/or liver diseases (18, 19).
Clinical disease occurs most frequently in patients who are homozygous null, SS and ZZ, and less frequently in patients who are heterozygous SZ or MZ. There are also variations in the speed of electrophoretic mobility of α1AT including fast (α1AT-F), medium (α1AT- M), slow (α1AT-S) and very slow (α1AT-Z). Table 3 summarizes the different phenotypes of α1AT (20).
The variants most frequently associated with liver disease are homozygous PiZZ, PiM and PiS and heterozygous PiSZ and PiMZ. Up to 15% of those born with homozygous PiZZ develop disease before the age of 20. The spectrum of liver disease manifested in the neonatal period with cholestasis and a clinical picture of neonatal hepatitis is very broad. They are one of the main causes of liver transplantation in this age group. Subsequently it presents as acute hepatitis or chronic cholestasis with decreased intrahepatic bile ducts and cirrhosis with or without hepatocellular carcinoma.
The mutant Z gene directs the synthesis of large amounts of mutant protein Z in the liver, and it accumulates within cells rather than being secreted into circulation. This accumulation within the hepatocytes causes liver damage through a cascade of hepatocellular apoptosis, regeneration and death (21).
If only 30% of homozygous patients progress to cirrhosis or liver failure, when should we suspect the presence of α1AT deficiency? The answer is in every child with neonatal cholestasis, in every patient with cirrhosis in childhood or adolescence, and whenever liver function tests show persistent alterations (22-25).
Histopathological findings are usually underestimated, but a correct diagnosis should be based on a proper correlation of clinical and laboratory studies including pathology and biopsy. In the first 12 weeks of life morphological alterations are subtle, so more than 80% of cases of α1AT deficiency are similar to INH. Consequently, a biopsy alone is insufficient to establish the diagnosis.
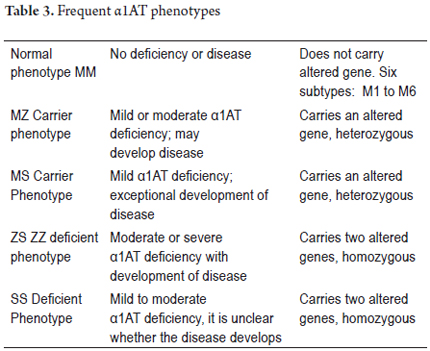
- Diagnosis is based on finding eosinophilic globules through histochemical and/or immunohistochemical studies. These globules are glycoproteins that have accumulated in the endoplasmic reticulum. They are PAS positive, resistant to diastase digestion and are located in the cytoplasm of hepatocytes in the periportal region. (Figure 8)
- Other findings are shared with other etiologies. Portal inflammation and fibrosis begin in the portal spaces until regeneration nodules or cirrhosis develops.
In infants under 12 or 13 weeks of age the number of α1AT globules may be insignificant, or they may not even be present, so diagnosis should aim at determining serum levels of α1AT and establishing its phenotype. The diagnostic accuracy PAS diastase stain (Pas-D) that is positive for globules is close to 99% for PiZ phenotypes in children with severe disease and close to 60% when the disease is moderately severe, but these globules cannot be found in less severe cases (1, 26).
DUCTOPENIA
Ductopenia includes a heterogeneous group of diseases characterized by the reduced number of small intrahepatic bile ducts. It has received other names such as hypoplasia and paucity of intrahepatic bile ducts.
One well-defined condition is known as Alagille syndrome or arteriohepatic dysplasia. It is associated with cardiac, facial, ocular and skeletal abnormalities but can also occur as a nonsyndromic entity that is frequently associated with other conditions. These include infections, especially CMV and rubella, metabolic diseases such as Alpha -1 antitrypsin deficiency, and chromosomal abnormalities such as Downs syndrome.
The pathogenesis of ductopenia is uncertain, although it is considered to be an entity that progressively destroys the bile ducts. Its evolution varies, and may it resolve spontaneously, produce fibrosis or even develop into cirrhosis.
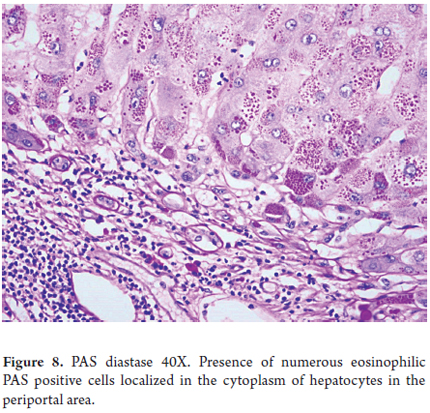
Diagnosis is based on finding marked diminution of the number of interlobular bile ducts, or even the absence of interlobular bile ducts (Figure 9). To establish the diagnosis the portal space ratio should be 0.4 or less (27, 28).
Other histopathological findings are shared with other entities that cause cholestasis including:
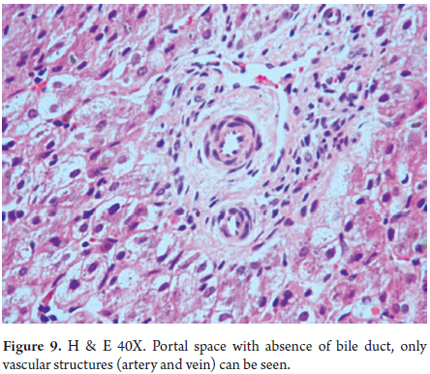
- Canalicular cholestasis (Figure 10).
- Small or not apparent portal spaces.
- Hepatocellular ballooning with occasional giant cell transformation.
- Extramedullary hematopoiesis foci.
- Initial Portal and periportal fibrosis of varying degree that may progress to cirrhosis.
CONCLUSION
Cholestatic disease in newborns and children are difficult to diagnose from both the clinical point of view and the histopathological point of view. They require analysis of a complete set of medical records, knowledge of family history, and imaging studies and usually require specialized laboratory, molecular biology and genetic studies. In some cases, diagnosis requires a liver biopsy to allow appropriate clinical correlation.
REFERENCES
1. Ovchinsky N, Moreira RK, Lefkowitch JH, Lavine JE. Liver biopsy in modern clinical practice: a pediatric point-of-view. Adv Anat Pathol 2012; 19(4): 250-62. [ Links ]
2. Feldman AG, Sokol RJ. Neonatal Cholestasis. Neoreviews 2013; 14(2). [ Links ]
3. Santos JL, Choquette M, Bezerra JA. Cholestatic liver disease in children. Curr Gastroenterol Rep 2010; 12(1): 30-9. [ Links ]
4. McKiernan PJ. Neonatal cholestasis. Semin Neonatol 2002; 7(2): 153-65. [ Links ]
5. Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr 2004; 39(2): 115-28. [ Links ]
6. Dehghani SM, Haghighat M, Imanieh MH, Geramizadeh B. Comparison of different diagnostic methods in infants with Cholestasis. World J Gastroenterol 2006; 12(36): 5893-6. [ Links ]
7. Sokol RJ, Mack C, Narkewicz MR, Karrer FM. Pathogenesis and outcome of biliary atresia: current concepts. J Pediatr Gastroenterol Nutr 2003; 37(1): 4-21. [ Links ]
8. Shinkai M, Ohhama Y, Take H, Kitagawa N, Kudo H, Mochizuki K, et al. Long-term outcome of children with biliary atresia who were not transplanted after the Kasai operation: >20-year experience at a childrens hospital. J Pediatr Gastroenterol Nutr 2009; 48(4): 443-50. [ Links ]
9. Pereira TN, Walsh MJ, Lewindon PJ, Ramm GA. Paediatric cholestatic liver disease: Diagnosis, assessment of disease progression and mechanisms of fibrogenesis. World J Gastrointest Pathophysiol. 2010; 1(2): 69-84. [ Links ]
10. Venigalla S, Gourley GR. Neonatal cholestasis. Semin Perinatol 2004; 28(5): 348-55. [ Links ]
11. Low Y, Vijayan V, Tan CE. The prognostic value of ductal plate malformation and other histologic parameters in biliary atresia: an immunohistochemical study. J Pediatr 2001; 139(2): 320-2. [ Links ]
12. Tatekawa Y, Asonuma K, Uemoto S, Inomata Y, Tanaka K. Liver transplantation for biliary atresia associated with malignant hepatic tumors. J Pediatr Surg 2001; 36(3): 436-9. [ Links ]
13. Vera A, Villaveces D, López R. Orthotopic liver transplantation for biliary atresia complicated by incidental cholangiocarcinoma. J Pediatr Gastroenterol Nutr 2012; 55(3): 336-7. [ Links ]
14. Moore L, Bourne AJ, Moore DJ, Preston H, Byard RW. Hepatocellular carcinoma following neonatal hepatitis. Pediatr Pathol Lab Med 1997; 17(4): 601-10. [ Links ]
15. Correa KK, Nanjundiah P, Wirtschafter DD, Alshak NS. Idiopathic neonatal giant cell hepatitis presenting with acute hepatic failure on postnatal day one. J Perinatol 2002; 22(3): 249-51. [ Links ]
16. Drut R, Gómez MA, Drut RM, Lojo MM. Human papillomavirus (HPV)-associated neonatal giant cell hepatitis (NGCH). Pediatr Pathol Lab Med 1996; 16(3): 403-12. [ Links ]
17. Domiati-Saad R, Dawson DB, Margraf LR, Finegold MJ, Weinberg AG, Rogers BB. Cytomegalovirus and human herpesvirus 6, but not human papillomavirus, are present in neonatal giant cell hepatitis and extrahepatic biliary atresia. Pediatr Dev Pathol 2000; 3(4): 367-73. [ Links ]
18. Kalsheker NA. alpha1-Antitrypsin deficiency: best clinical practice. J Clin Pathol 2009; 62(10): 865-9. [ Links ]
19. Zarrilli F, Elce A, Scorza M, Giordano S, Amato F, Castaldo G. An update on laboratory diagnosis of liver inherited diseases. Biomed Res Int 2013. [ Links ]
20. Stoller JK, Aboussouan LS. Alpha1-antitrypsin deficiency. Lancet 2005; 365(9478): 2225-36. [ Links ]
21. Teckman JH, Jain A. Advances in alpha-1-antitrypsin deficiency liver disease. Curr Gastroenterol Rep 2014; 16(1): 367. [ Links ]
22. Chappell S, Hadzic N, Stockley R, Guetta-Baranes T, Morgan K, Kalsheker N. A polymorphism of the alpha1-antitrypsin gene represents a risk factor for liver disease. Hepatology 2008; 47(1): 127-32. [ Links ]
23. Perlmutter DH, Brodsky JL, Balistreri WF, Trapnell BC. Molecular pathogenesis of alpha-1-antitrypsin deficiency-associated liver disease: a meeting review. Hepatology 2007; 45(5): 1313-23. [ Links ]
24. Campbell KM, Arya G, Ryckman FC, Alonso M, Tiao G, Balistreri WF, et al. High prevalence of alpha-1-antitrypsin heterozygosity in children with chronic liver disease. J Pediatr Gastroenterol Nutr 2007; 44(1): 99-103. [ Links ]
25. De Serres FJ. Alpha-1 antitrypsin deficiency is not a rare disease but a disease that is rarely diagnosed. Environ Health Perspect 2003; 111(16): 1851-4. [ Links ]
26. Topic A, Prokic D, Stankovic I. Alpha-1-antitrypsin deficiency in early childhood. Fetal Pediatr Pathol 2011; 30(5): 312-9. [ Links ]
27. Alagille D. Estrada A, Hadchouel et al. Syndrome paucity of interlobular bile ducts Alagille syndrome of arteriohepatic dysplasia): Review of 80 cases. The Journal of Pediatrics 1987; 110 (2): 195-200. [ Links ]
28. De Tommaso AM, Kawasaki AS, Hessel G. Paucity of intrahepatic bile ducts in infancy. Experience of a tertiary center. Arq Gastroenterol 2004; 41(3): 190-192. [ Links ]
