










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.30 no.3 Bogotá July/Sept. 2015
Acute liver failure due to hepatitis B: a case report
Ana Madeleine Barrera L. (1), Laura Daniela Rincón (2), Fernando Peñaloza Cruz MD. (3), Lina Patricia Vargas N. (4), Farhi Alonso Delgado Medina (5)
(1) Internal Medicine Resident at the Universidad del Rosario and Hospital Occidente de Kennedy.
(2) Medical Student, Internal Medicine Rotation at the Hospital Occidente de Kennedy.
(3) Internist and Gastroenterologist at the Hospital Occidente de Kennedy.
(4) Internal Medicine Resident at the Universidad del Rosario and Hospital Occidente de Kennedy.
(5) Internist at the Hospital Occidente de Kennedy.
Received: 22-11-13 Accepted: 21-07-15
Abstract
We report the case of a 37 year old woman who came to the hospital because of jaundice and a fever. Her symptoms were associated with significant liver impairment and a necroinflammatory pattern due to viral hepatitis B although she had no relevant medical history. Her symptoms developed rapidly until death. We present the factors that may have influenced her progression to fulminant liver failure as described in the literature.
Keywords
Acute liver failure, causes of hepatitis B, hepatitis B virus, hepatitis B viral mutations, hepatitis B therapy.
INTRODUCTION
Fulminant hepatic failure is a severe and acute injury which presents infrequently. It is probably related to or induced by an exaggerated immune response to viral hepatitis, or as described in most of the series, to acetaminophen poisoning and acute liver necrosis.
It is typically described as encephalopathy occurring after a prolonged time (INR> 1.5). Depending on the amount of time of evolution between symptoms and the development of encephalopathy, it is classified into either hyperacute, acute or subacute types.
Since it is a rare entity, there are no large-scale studies describing its natural history, or level of evidence for management. Nevertheless, in 2011 the American Association for the Study of Liver Diseases (AASLD) presented a background review of expert opinions of the document published in 2005 regarding diagnosis, treatment and transplant expectations.
With this article, we want to put constraints in the early diagnosis of the etiology of liver failure, as well as its proper handling, in context. In addition, we briefly review the subject with emphasis on factors that influence the evolution of hepatitis B infections into fulminant hepatic failure.
CASE DESCRIPTION
The patient was a 37 year old woman who had no relevant medical history. She was born in Ibague, Tolima but lived in Bogota DC. She came to the hospital after four 4 days of diffuse abdominal pain associated with subjective fever and jaundice. Physical examination showed the following information: Blood pressure 120/70, heart rate 80 per min, respiratory rate 18 per min, blood oxygen saturation 93%, FiO2 0.21, jaundice, no shortness of breath, no jugular venous distension, normal cardiopulmonary, painful epigastric palpation, painful right upper quadrant palpation, no peritoneal irritation, no signs of chronic liver disease, no signs of neurological distress and no encephalopathy.
Paraclinical tests showed the following information: Liver profile of necroinflammatory pattern, total bilirubin 7.58 mg/dl, direct bilirubin 5.97 mg/dl, alanine aminotransferase (ALT) 6.315 mg/dl, aspartate transaminase (AST) 8.947 mg/dl, PT 24, INR 1.8, blood glucose 114 mg/dl, electrolytes with hyponatremia (130 Na, 99.4 CL, 3.65 K), renal function not compromised (creatinine 0.56, bun 14.7). A sonogram of the liver was suggestive of hepatitis.
The patient was admitted for clinical observation because of the high risk of liver failure and the need to study possible liver disease. Fluid resuscitation was started, and studies of liver disease began with a viral profile (HBsAg, HVC, Total HVA, IgM Hep E, IgG, M Varicella zoster), an immunological profile (Anas - ASMA), and levels of ceruloplasmin and copper. The patient and family were interviewed and did not refer to any history of alcoholism, so this source was not considered likely. Similarly they did not mention any ingestion of toxic substances, homeopathic medicines or excessive use of acetaminophen.
During the first three days in the hospital, the patient remained hemodynamically stable without signs of hepatic encephalopathy, but with worsening of liver function. Tests for HBsAg were frankly positive. A hepatitis virus panel of tests for HBeAg, Anti HBeAg, Anti-Hepatitis B core total antibodies, and Anti-Hepatitis D antibodies was requested to assess whether the patient had an acute and/or a reactivated chronic infection. Treatment began with 0.5 mg doses of entecavir daily. Patient was referred for liver transplantation. The patient's LDH level was elevated as can be seen in Table 1. This condition has been described in the prodromal period of hepatic failure the literature. It reaches its maximum level with the onset of jaundice in viral hepatitis. Its progression in relation to hepatic function can be seen in Figure 1.
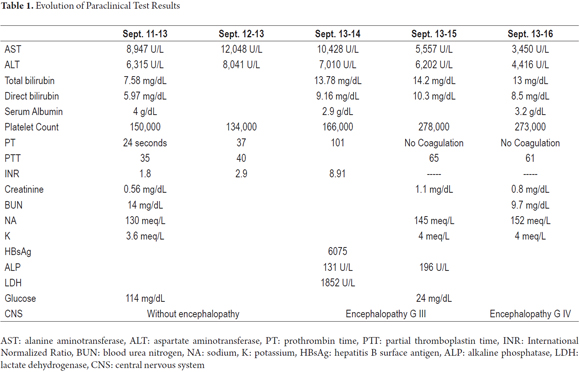
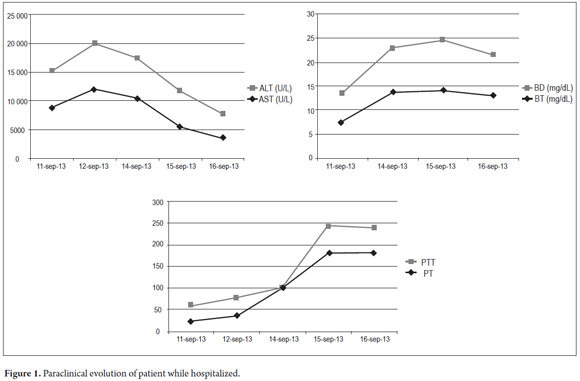
On the fourth day the patient suffered overall deterioration to grade II hepatic encephalopathy. Management metronidazole, lactulose (to counter encephalopathy) and N-acetylcysteine to manage liver disease was begun. Arterial blood gases showed an elevated anion gap which was related to a high level of high metabolic acidosis due to liver failure (FiO2: 0.28, pH: 7.47, pCO2: 15, PO2: 48, HCO3: 10.9, SatO2: 86%). This was treated medically with bicarbonate of soda. The patient was moved to the intensive care unit for progression to hepatic encephalopathy grade III and hemodynamic deterioration requiring vasopressor support. She did not respond favorably, and on the 5th day she developed grade IV hepatic encephalopathy. Orotracheal intubation became necessary to protect respiration. A CT scan of her brain was taken (Figure 2) and appeared to be normal. The fundi of her eyes were without papilledema, but she was given a bolus of mannitol because of the possibility of intracranial hypertension. There was no clinical improvement. She developed supraventricular tachycardia and hemodynamic collapse requiring electrical and pharmacological cardioversion. She had also developed upper gastrointestinal bleeding for which infusion of omeprazole was initiated. She was transfused with 15 cc/kg of plasma plus vitamin K. The patient progressed to cardiac arrest. She was unresponsive to advanced maneuvers and died 5 days after admission to the institution and nine days after onset of symptoms. The cause of death was determined to be fulminant liver failure secondary to hepatitis B since other major studios required referrals to a hospital of greater complexity which were not possible to get in time because this is a state hospital.
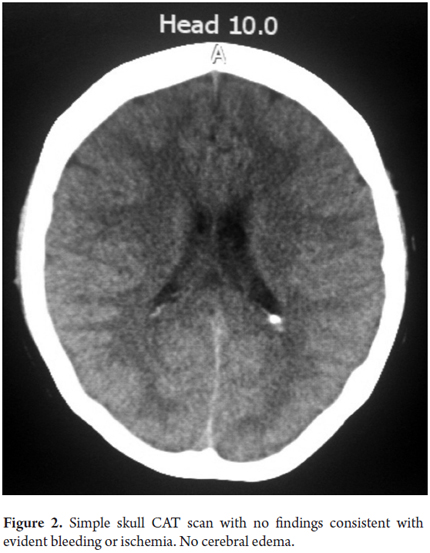
DISCUSSION
Liver failure is defined as abnormality of coagulation (INR> 1.5) associated with any degree of encephalopathy which quickly progresses in a patient without preexisting cirrhosis or liver disease within 26 weeks (Table 2) (1, 2). This describes our patient.

Epidemiologically, the incidence of this entity in Colombia is unknown. Nevertheless, other cases have been reported. One reported case was secondary to autoimmune hepatitis in a patient treated with pegylated interferon for chronic infection with hepatitis C, and another was a patient suffering from and autoimmune hepatitis (3, 4).
In the US and Europe, most cases are secondary to toxic causes, the principal of which is acetaminophen poisoning (46%). This is followed by viral causes (12%), the dominant of which is hepatitis B virus (7%) (5). Fulminant liver failure due to hepatitis B can occur from the onset of acute infection, from aggravation of a chronic infection or coinfection with Hepatitis C or D, or from Hepatitis E in 15% to 25% of pregnant women especially in the third trimester of pregnancy (6-9). Other causes include autoimmune conditions and drugs other than acetaminophen such as amiodarone, quetiapine and valproic acid (3, 10-13).
Fulminant liver failure due to HBV is rare, but it has a high mortality rate and poor prognosis for the patient. The mortality rate is even higher when there is co-infection with hepatitis D virus (HDV). A large inoculum is associated with a shorter incubation period than with other fulminant liver failure. In contrast, a quick immune response is more closely related to the development of fulminant liver failure than to increased viral replication (14). This means that the early turn of HBsAg to anti-HBs might be involved in the pathogenesis of fulminant liver failure (15). In addition, the association between a mutation of HBV and the development of fulminant hepatic failure has previously been reported (16, 17). A mutation has been described in the precore, core and protein of HBV that apparently induces hepatocyte apoptosis. Nevertheless, it has not been documented that it is a prerequisite for the development of fulminant liver failure (18, 19).
Pathophysiology is uncertain about how this mutation produces massive hepatocellular damage or about how it leads to a b immune response by T lymphocytes. Post mortem examination evidence shows the liver suffers massive necrosis. It is believed that this mutation in the precore either causes massive T cell activation or causes massive damage to the hepatocytes by a rapid increase of HBcAg (15).
Another theory involves alteration of endothelial nitric oxide (ONe). Its normal function is vasodilation for hemodynamic control. When its levels are decreased and nitric oxide synthase (ONS) increases, it causes a change in vasodilation and considerably increases toxic free radical production resulting in damage to the endothelium. In addition, it causes massive activation of caspases leading to large scale apoptosis (20). In fulminant liver failure, Kupffer cells in the sinusoids produce nitric oxide allowing for increased vascular permeability and thus increased concentrations of macrophages in the liver. Furthermore, there is less expression of endothelial nitric oxide (eNO) in the bile ducts, endothelial cells and lymphocytes. Additionally, at the onset of fulminant liver failure, serum levels of C-reactive protein (CRP) are normal, but before the final stages of fulminant liver failure CRP serum levels can be as high as 10 mg/dl. Levels rise until the liver loses its capacity of protein synthesis and PCR begins to decrease (21). Levels of coagulation factors such as factor V, protein C and protein S associated with elevated serum levels of bilirubin also decrease. Levels of these clotting factors were not measured, but incalculable clotting times first added to rising bilirubin and initially elevated transaminases and then descended in parallel to increase clotting times and bilirubin levels. This provided pathophysiological evidence of the progression of liver failure. Also, CRP levels may be a predictor of progression and severity of fulminant liver failure (22). This is the basis for the idea about treatment with prostacyclin, however this has not yet proven to be beneficial. If in fact it is not, it could increase the risk of complications (23). In any case, we did not have the capacity to measure CRP at the time that the patient studied here was treated, so we do not have these data to present.
There are various classifications based on the time between the onset of jaundice and development of encephalopathy which also provide clues about the cause of the disease, probable complications and prognosis with treatment according to the interval (Figure 3) (24). Independent of any limitations in terms of proper diagnosis and management, the time between the onset of jaundice and development of encephalopathy in our patient was seven days which makes this a case of acute or fulminant liver failure.

All of these patients must be handled in the ICU in order to stabilize and control the complications arising from fulminant liver failure and as a bridge to liver transplantation. In most situations transplantation is the only effective treatment (25). Meanwhile, the tests listed in Table 3 should be conducted and management should focus on problems, taking into account predictors of poor prognosis as guides for priority selection for transplantation (6).
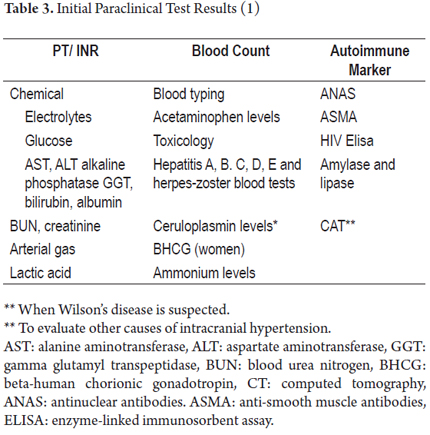
Encephalopathy
Cerebral edema occurs most in the acute presentation of fulminant liver failure and is the leading cause of mortality in these patients. The first link in the management of these patients is to prevent and manage intracranial hypertension (ICH). For patients with Grade 1 and Grade 2 hepatic encephalopathy it is advisable to minimize stimuli and maintain the head elevated at 30°. For patients with Grades 3 and 4 of hepatic encephalopathy orotracheal intubation is, as in our patient, probably necessary. These patients most likely will be suffering from severe intracranial hypertension (ICP> 25 mm Hg for> 5 minutes, or ICP> 40 mm Hg, or cerebral perfusion pressure <40 mm Hg for more than two hours). Mannitol IV bolus at doses of 0.25 to 1.0 g/kg is recommended for these patients when the osmolarity is less than 320 mOsm/L (1). This was administered to patient, despite our inability to monitor intracranial pressure.
Inducing mild hypernatremia of 145 to 150 mmol/L can reduce the incidence and severity of ICH (26, 27). Inducing therapeutic hypothermia (33°C-35°C) may be beneficial for reducing splanchnic production of ammonia, restoring autoregulation of cerebral hemodynamics and decreasing oxidative metabolism in the brain (28).
Hyperventilation (PaCO2 of 25 to 30 mmHg) should only be used when there is acute neurologic deterioration (ICP> 20 mmHg with evidence of increased cerebral blood flow).
Liver Disease / Coagulopathy
Spontaneous bleeding occurs in less than 10% of cases. It is secondary to liver failure and increased consumption of procoagulant factors. In the absence of bleeding, and when there is no need for invasive procedures (24), prophylactic correction of coagulopathy may not reduce the risk of significant bleeding and can even exacerbate volume overload. Consequently, it is recommended only when an invasive procedure is necessary or when there is bleeding as occurred in our patient. She received plasma + Vitamin K plus the usual management for upper gastrointestinal bleeding.
N-acetyl cysteine (29) has been studied in relation to hepatic acetaminophen poisoning, but its use is also for maintaining infusion until hepatic encephalopathy is resolved and until INR levels fall. The recommended dose is 150 mg/kg via IV bolus in 15 to 60 minutes followed by maintenance infusion of 5.12 mg/kg/h for four hours, and then 6.25 mg/kg/h.
Life Support
Life support must ensure a state of euvolemia. If resuscitation is required, you must start with 0.9% normal saline solution, stagger vasopressors (norepinephrine) to maintain MAP> 65 mmHg, and target central venous pressure at 6 to 10 mm Hg (1). Acute renal failure occurs in 50% of cases, indicating renal replacement therapy in special situations (Table 4).

The prophylactic use of antibiotics is not indicated except in case of progression to hepatic encephalopathy, refractory hypotension, systemic inflammatory response syndrome (SIRS) or in preparation for a liver transplant. This was the case of our patient (30).
Transplantation (table 5)
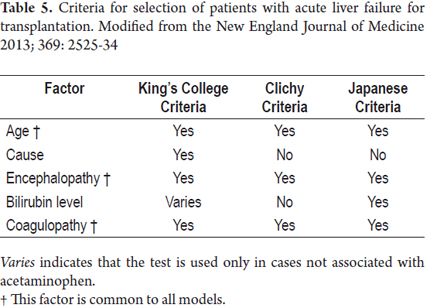
Transplantation is the only treatment option for some specific causes of acute liver failure (24). Nevertheless, it is not available for every patient. In fact, less than 10% of liver transplantations are performed in patients with acute liver failure. Survival rates for these patients are always lower than those associated with elective liver transplantation. This is related to infections during the first three months after surgery. The risk is higher among older recipients and among those who received grafts from donors whose ABO blood group is different from that of the recipient.
CONCLUSIONS
The case described in this study is that of a young woman with no history of liver disease, no known risk factors for liver disease, no known ingestion of toxic substances but who developed a clinical picture of acute fulminant liver failure. Because of the administrative limitations of this state hospital, we cannot determine whether the clinical course of the patient was related to hepatitis B infection, whether it was an acute episode or a relapse, or whether there was co-infection with hepatitis B which the literature describes as the most common presentation. Neither can we rule out other etiologies because studies that would allow this require prior authorization and referral. Since results could not be obtained, it was not possible to refer this patient to a liver transplant center.
With this case, we have tried to place this important issue in its medical context, but also in the context of the administrative deficit of our social institutions. They have been forgotten by the state and the financial resources they need have been diverted elsewhere. The results are delays in timely access, failure to modernize diagnostic support, limited access to complex hospitals, and limited access to the private institutions with the capacity to perform liver transplantation.
REFERENCES
1. Umaña García P. Manejo de la falla hepática fulminante - Experiencia en la Fundación Santa Fe de Bogotá. Acta Med Col. 1990;15(1). [ Links ]
2. Lee WM. Uptodate HEPATOLOGY The Management of Acute Liver Failure AASLD Position Paper. [Online].; 2012 [cited 2013 Nov. [ Links ]].
3. Correa Gaviria S. Reporte de un caso de falla hepática aguda por hepatitis autoinmune en una paciente en tratamiento con interferón pegilado para infección crónica por el virus de la hepatitis C. Rev Col Gastroenterol. 2013;28(1):69-73. [ Links ]
4. Carvajal C. Falla hepática aguda, caso clínico y revisión de la literatura. Acta Col Cuidado Intensivo. 2011;11(4):301-9. [ Links ]
5. Arata S. Hepatic failure in pregnancy successfully treated by online hemodiafiltration: Chronic hepatitis B virus infection without viral genome mutation, case report. Hepatol Res. 2013; doi: 10.1111/hepr.12090. [ Links ]
6. ICHAI P,SD. Etiology and prognosis of fulminant hepatitis in adults. liver transplant. 2008;14(10): Suppl 2. [ Links ]
7. Mishra N, Walimbe AM, Arankalle VA. Hepatitis E virus from India exhibits significant amino acid mutations in fulminant hepatic failure patients. Virus Genes. 2013;46:47-53. [ Links ]
8. Devi Salam G. Serum tumor necrosis factor-alpha level in hepatitis E virus-related acute viral hepatitis and fulminant hepatic failure in pregnant women. Hepatol Res. 2013;43:826-835. [ Links ]
9. Jayakumar S. Fulminant viral hepatitis. Crit Care Clin. 2013;29(2013)677-97. [ Links ]
10. Nasser M. Hyperacute drug-induced hepatitis with intravenous amiodarone: Case report and review of the literature. Drug Healthc Patient Saf. 2013;5:191-8. [ Links ]
11. Fink DL BE. Liver transplantation for acute liver failure due to efavirenz hepatotoxicity: The importance of routine monitoring. Int J STD AIDS. 2013;24(10):831-3. [ Links ]
12. Mettananda S. Posterior reversible encephalopathy syndrome in a survivor of valproate-induced acute liver failure: A case report. J Med Case Rep. 2013;7(1):144. [ Links ]
13. Al Mutairi F. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Reports. 2012;6:418. [ Links ]
14. Liang TJ, Hasegawa K, Rimon N, et al. A hepatitis B virus mutant associated whit an epidemic of fulminant hepatitis. N Engl J Med. 1991;324(24):1705-9. [ Links ]
15. Trepo CG, Robert D, Motin J, et al. Hepatitis B antigen (HBSAg) and/or antibodies (anti-HBS and anti-HBC) in fulminant hepatitis: Pathogenic and prognostic significance. Gut. 1976;17:10-13. [ Links ]
16. Kitano K, Kobayashi H, Hanamura M, et al. Fulminant hepatitis after allogenic bone marrow transplantation caused by reactivation of hepatitis B virus with gene mutations in the core promotor region. Eur J Haematol. 2006;77:255-8. [ Links ]
17. Khare S, Negi SS, Singh S, et al. Genetic analysis of precore/core and partial pol genes in an unprecedented outbreak of fulminant hepatitis B in India. Epidemiol Infec. 2012;140(10):1823-9. [ Links ]
18. Rivero M, Crespo J, Fábrega E, et al. Apoptosis mediated by the Fas system in the fulminant hepatitis by hepatitis B virus. J Viral Hepatitis. 2002;9(2):107-13. [ Links ]
19. Omata M, Ehata T, Yokosuka T, et al. Hepatitis B virus mutation and fulminant hepatitis. N Engl J Med. 1991;324(24):1699-1704. [ Links ]
20. Leifeld L, Fielenbach M, Dumolin FL, et al. Inducible nitric oxide synthase (iNOS) and endothelial nitric oxide synthase (eNOS) expression in fulminant hepatic failure. J Hepatol. 2002;37(5):613-9. [ Links ]
21. Silvestre JP. Impact of fulminant hepatic failure in C-reactive protein? J Crit Care. 2010;25(4)657:e7-12. [ Links ]
22. Chu CJ, Hsiao CC, Wang TF, et al. Prostacyclin inhibition by indomethacin aggravates hepatic damage and encephalopathy in rats with thioacetamide-induced fulminant hepatic failure. World J Gastroenterol. 2005;11(2):232-6. [ Links ]
23. Davenport A. Adverse effects on cerebral perfusion of prostacyclin administered directly into patients with fulminant hepatic failure and acute renal failure. Nephron. 1991;59(3):449-54. [ Links ]
24. Bernal W. Acute liver failure: Review article. N Engl J Med. 2013;369:2525-34. [ Links ]
25. Gómez Cabeza de Vaca V. Liver transplantation due to fulminant hepatic failure. Transplant P. 2012;44:2076-7. [ Links ]
26. Murphy N. The effect of hypertonic sodium chloride on intracranial pressure in patients with acute liver failure. Hepatology. 2004;39(2):464-70. [ Links ]
27. Bernal W. Intensive care management of acute liver failure. Semin Liver Dis. 2008;28(2):188-200. [ Links ]
28. Stravitz RT. Therapeutic hypothermia for acute liver failure. Crit Care Med. 2009;37(Suppl 7):S258-64. [ Links ]
29. Hu J, Zhang Q, Ren X, et al. Efficacy and safety of acetylcysteine in «non-acetaminophen» acute liver failure: A meta-analysis of prospective clinical trials. Clin Res Hepatol Gastroenterol. 2015;S2210-7401(15)00031-5. [ Links ]
30. Stravitz RT. Intensive care of patients with acute liver failure: Recommendations of the U.S. Acute Liver Failure Study Group. Crit Care Med. 2007;35(11):2498-508. [ Links ]
