










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957
Rev Col Gastroenterol vol.30 no.4 Bogotá out./dez. 2015
Case Report of Syndromic Biliary Atresia in a Pediatric Patient
Patricia Ruíz N. MD. (1), Karen Aguirre R. MD. (1), Catalina Mesa M. MD. (2), Lilian Lara L. MD. (2)
(1) Specialist in pediatric gastroenterology and hepatology at the Hospital Pablo Tobon Uribe in Medellín, Colombia.
(2) Pediatric Resident at the Hospital Pablo Tobon Uribe in Medellín, Colombia.
Received: 06-09-14 Accepted: 20-10-15
Abstract
Biliary atresia is an obstructive neonatal cholangiopathy of unknown etiology that produces damage to the parenchyma of the liver and to the intrahepatic and extrahepatic bile ducts. It is the most common cause of neonatal cholestasis and liver transplantation in the pediatric population. In most cases it manifests as an isolated malformation. This article presents the case of a 75 day old patient with biliary atresia associated with abdominal heterotaxy. Biliary atresia syndrome together with splenic malformation has been previously described, as have biliary atresia associated with anatomical malformations of the spleen, pancreas, and heart. It occurs with genitourinary malformations less frequently. The prognosis of patients with biliary atresia has significantly improved with early recognition of signs and symptoms and timely performance of hepatoportoenterostomy (Kasai portoenterostomy).
Keywords
Biliary atresia, visceral heterotaxy
INTRODUCTION
Biliary atresia is an obstructive neonatal cholangiopathy caused by an inflammatory process that causes progressive fibrosis and obliteration of the intrahepatic and extrahepatic bile ducts. It is associated with damage to the parenchyma of the liver (1). It is the most common cause of neonatal cholestasis and biliary obstruction and occurs in 1 in 15,000 to 20,000 live births. If left untreated, the resulting cholestasis leads to cirrhosis and liver failure (2, 3). Although in most cases, biliary atresia occurs alone, 10% to 20% of patients have associated vascular and visceral malformations (3, 4).
Early performance of Kasai Portoenterostomy has a significant impact on the survival of patients with biliary atresia, so primary health care personnel need to be able to use practical tools such as clinical histories so that a multidisciplinary team specializing in hepatology can make early diagnosis and evaluation (5).
This article reports the case of a pediatric patient with biliary cirrhosis and portal hypertension secondary to syndromic biliary atresia, explains the indications for liver transplantation in these patients and emphasizes the value of semiotics for early diagnosis and treatment in cases of neonatal cholestasis which can enable timely interventions that impact positively on the prognoses of these patients.
CLINICAL CASE
The patient was a 75-day old boy whose mother had suffered gestational diabetes which did not require pharmacological management. There was no history of consanguinity in the parents, and the baby's weight and size were normal for his gestational age. Neonatal adaptation had been spontaneous. Fifteen days after he was born, the baby's mother brought him to the pediatric hepatology clinic because of acholia. He had been to primary level health providers twice where he had been given phototherapy.
Fifty days after birth, persistence of symptoms together with increased waist circumference but unchanged weight led another visit to the pediatric hepatology clinic. The child's weight was 5.85 kilograms and his length was 57 centimeters. A grade 2/4 midsystolic murmur was found together with ascites, collateral circulation, hepatomegaly, splenomegaly, a protruding umbilical hernia and large bilateral inguinal hernias. Paraclinical tests showed hypoalbuminemia, cholestasis, and altered coagulation and liver enzymes (see Table 1).
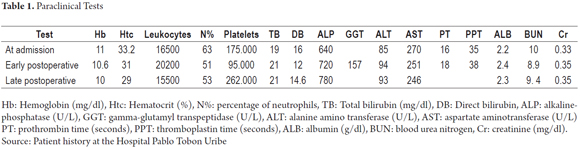
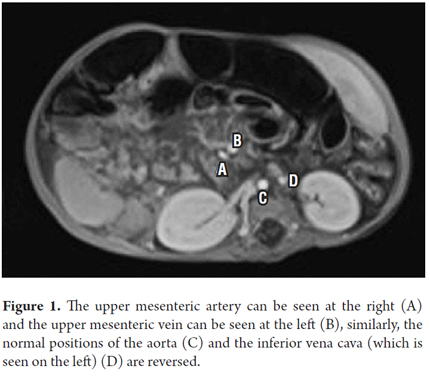
An echocardiogram was normal. Abdominal MRI showed a prominent four millimeter hepatic artery branching from the celiac artery, a permeable portal vein branching from the superior mesenteric vein and located on the left side, a superior mesenteric artery on the right side, the retrohepatic vena cava, and agenesis of the inferior vena cava (see Figure 1). The left central liver was enlarged with heterogeneous changes due to chronic liver disease with multiple venous collaterals and seven structures compatible with polysplenia. The stomach was on the right side (see Figure 2). Testicular ultrasound showed a gigantic inguinal hernia on the right side, large hydroceles on both sides bilateral and testicles in the scrotum. A liver biopsy found canalicular and cytoplasmic cholestasis, Kupffer cells, and numerous giant multinucleated cells formed by fusion of hepatocytes. Biochemical tests showed thyroid function and immunoglobulins within normal limits and alpha fetoprotein of 66.53. Infection by hepatotropic viruses and HIV were ruled out. Liver and biliary scintigraphy showed biliary atresia. A Kasai procedure was performed when the patient was 75 days old. Two months after the intervention, the initial symptoms persisted. The patient's weight was 7 kg, his length was 58 centimeters. The very small weight/height gain is associated with a PELD (pediatric end-stage liver disease) score of 20 points, so the patient was considered to be a candidate for liver transplantation.
DISCUSSION
Biliary atresia is the main cause of cholestasis and the most common cause of liver transplantation in new born infants (6). Biliary atresia without other anatomical malformations accounts for 80% of cases, but the remaining 20% have various other anatomical malformations such as polysplenia, portal vein abnormalities, intestinal malrotation, and absence of the inferior vena cava. In addition, they are likely to have cardiac and pancreatic disorders (1).
Davenport et al. (3) have described the biliary atresia syndrome and splenic malformation which they identified as a subset with a distinct etiology and worse prognosis. In this syndrome, biliary atresia coexists with extrahepatic malformations of which the most common is polysplenia which may in turn be associated with thoracoabdominal heterotaxy such as intestinal malrotation and vascular disorders.
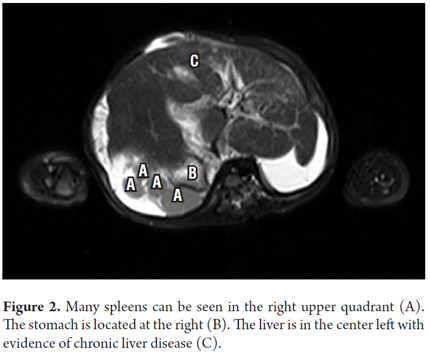
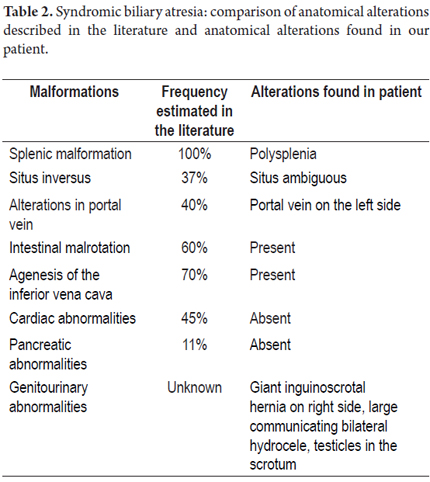
Although the cause of biliary atresia is unknown, the obliteration of bile ducts implies that there are genetic, infectious and inflammatory triggers. The consistency of the anatomical abnormalities together with biliary atresia implies a specific event in embryogenesis and suggests a genetic component (1). The possible pathophysiological mechanism is an alteration in the process of laterality during embryogenesis which result in defective development of organs in the abdominal cavity and thorax, failure to develop these organs, or transposition of these organs (7). According to the situs classification (See Figure 3) (8), the patient is a case of ambiguous situs with polysplenia given by the position of the stomach on the right side, inferior vena cava on the left side, intestinal malrotation and associated polysplenia. No pancreatic and cardiac disorders were demonstrated in our case. (see Table 2). Although there was no clear association with genitourinary abnormalities such as bilateral hydrocele in this patient, M. Salih Deveci reported a case of biliary atresia and splenic malformation with congenital hydrocele (9).
Three studies have reported that there seems to be a relationship between syndromic biliary atresia in babies and gestational diabetes found in the mother as was the case with our patient.
The prognosis for portoenterostomy depends on many factors some of which cannot be changed such as histology of the liver, portal pressure and polysplenia. One of the factors that can be modified to improve the patient's prognosis is age at surgery. Serinet Wildhaber et al. (10) report that the 2-year survival rate of the native liver is 65.5% when surgery is performed between 31 and 45 days after birth, 57.1% between 61 and 75 days after birth, and only 42% when surgery is performed more than 90 days after birth. In the case we describe, the portoenterostomy was performed at 75 days after birth, due to late admission.
Tien Hau Lien and colleagues (11) used a screening test with cards containing the colors of the stool. The results of this screening program were amazing: the study reported a 65.7% rate for portoenterostomy% in patients younger than 60 days who had access to the screening program using the cards with the colors of feces, while the group that did not have the screening program obtained a rate of 49.4 %. Implementation of this type of screening program can achieve better prognoses in the short and long term.
As mentioned above, biliary atresia is the most common indication for liver transplantation (12). In cases of this disease, especially in the clinical case presented, an ineffective portoenterostomy and signs of liver failure were considered indications for entering the patient into a liver transplant protocol. After portoenterostomy, our patient failed to restore bile flow and continued with progressive cholestasis, impaired liver function and poor weight and height gain (see Table 1). The survival rate of patients with biliary atresia alone following liver transplantation is approximately 90%, however the presence of complex vascular abnormalities and other extrahepatic defects is associated with more complications and reduced post-transplant survival (12).
In the Hospital Pablo Tobón Uribe which is a reference center for liver transplantation, 23 patients with biliary atresia were admitted between January 2005 and June 2014. Only one patient, a clinical case discussed in a previously published paper, had been diagnosed with syndromic biliary atresia. The median age at admission was 3 months. Only four of the 23 patients admitted had been referred by 1st and 2nd level health care centers which suggests that most of these patients were referred from centers of greater complexity of care but without trained management personnel. This may have delayed specific interventions and lessened the prognoses of these patients.
CONCLUSIONS
Syndromic biliary atresia is a rare condition. Patients with this condition have worse prognoses than do patients with biliary atresia alone. Early diagnosis of this disease to detect possibly associated anomalies and prevent liver failure is essential.
The mainstay of treatment for this pathology is timely Kasai portoenterostomy. In the event that this is not done or is not successful, liver transplantation becomes a necessity.
REFERENCES
1. Hartley JL, Davenport M, Kelly DA. Biliary atresia. The Lancet. 2009;374(9702):1704-13. [ Links ]
2. Feldman AG, Sokol RJ. Neonatal Cholestasis. NeoReviews. 2013;14(2):e63-73. [ Links ]
3. Davenport M, Tizzard SA, Underhill J, Mieli-Vergani G, Portmann B, Hadić N. The biliary atresia splenic malformation syndrome: A 28-year single-center retrospective study. J Pediatr. 2006;149(3):393-400. [ Links ]
4. Tanano Hirofumi, Akira Okada. Biliary atresia Associated With Congenital Structural Anomalies. J Pediatr Surg. 1999;34(11):1687-90. [ Links ]
5. Moyer V, Freese DK, Whitington PF, Olson AD, Brewer F, Colletti RB, et al. Guideline for the evaluation of cholestatic jaundice in infants: recommendations of the North American Society for Pediatric Gastroenterology, Hepatology and Nutrition. J Pediatr Gastroenterol Nutr. 2004;39(2):115-28. [ Links ]
6. C. Díaz Fernández, E. Frauca Remacha. Atresia biliar: una enfermedad grave. Arch Argent Pediatr. 2014;112(6):542-7. [ Links ]
7. Peeters H, Devriendt K. Human laterality disorders. Eur J Med Genet. 2006;49(5):349-62. [ Links ]
8. Corral G, Labra A, Schiappacasse G. Manifestaciones abdominales de las anomalías del Situs Ambiguous en el adulto: A propósito de cuatro casos. Rev Chil Radiol. 2013;19(1):38-43. [ Links ]
9. Deveci MS, Deveci G. Biliary atresia splenic malformation syndromeIs it a result of embryonically midline rotational defects? A case report. J Pediatr Surg. 2000;35(9):1377-80. [ Links ]
10. Serinet M-O, Wildhaber BE, Broue P, Lachaux A, Sarles J, Jacquemin E, et al. Impact of Age at Kasai Operation on Its Results in Late Childhood and Adolescence: A Rational Basis for Biliary Atresia Screening. Pediatrics. 2009;123(5):1280-6. [ Links ]
11. Lien TH, Chang M-H, Wu J-F, Chen H-L, Lee H-C, Chen A-C, et al. Effects of the infant stool color card screening program on 5-year outcome of biliary atresia in taiwan. Hepatology. 2011;53(1):202-8. [ Links ]
12. C. Díaz Fernández, E. Frauca Remacha. Trasplante hepático pediátrico: indicaciones, técnicas quirúrgicas, complicaciones y tratamiento. An Pediatría. 2004;60(1):42-55. [ Links ]
