










Serviços Personalizados
Journal
Artigo
 texto em
texto em  Inglês (pdf)
Inglês (pdf)
 Artigo em XML
Artigo em XML Referências do artigo
Referências do artigo
 Enviar este artigo por email
Enviar este artigo por emailIndicadores
-
 Citado por SciELO
Citado por SciELO -
 Acessos
Acessos
Links relacionados
-
 Citado por Google
Citado por Google -
 Similares em
SciELO
Similares em
SciELO -
 Similares em Google
Similares em Google
Compartilhar

 Permalink
PermalinkRevista colombiana de Gastroenterología
versão impressa ISSN 0120-9957
Rev Col Gastroenterol vol.30 no.4 Bogotá out./dez. 2015
Telangiectasia hemorrágica hereditaria. Reporte de caso
A Case Report of Hereditary Hemorrhagic Telangiectasia (HHT)
Martín Alonso Gómez MD. (1), Oscar Fernando Ruiz MD. (2), William Otero MD. (3)
(1) Profesor asociado de Gastroenterología, Facultad de Medicina, Universidad Nacional. Gastroenterólogo Hospital El Tunal. Gastroenterólogo UGEC. Bogotá, Colombia.
(2) Internista y Gastroenterólogo. Universidad Nacional de Colombia. Gastroenterólogo UGEC y Hospital El Tunal. Bogotá, Colombia.
(3) Profesor de Medicina. Gastroenterólogo, Unidad de Gastroenterología. Universidad Nacional de Colombia. Clínica Fundadores, Bogotá, Colombia.
Fecha recibido: 26-02-15 Fecha aceptado: 20-10-15
Resumen
La telangiectasia hemorrágica hereditaria o síndrome de Osler-Weber-Rendu es una entidad autosómica dominante rara, con una frecuencia que oscila entre 1 por cada 16 300 habitantes a 1 por cada 1331 dependiendo de la población y la localización geográfica, sin predilección de género. Se caracteriza clínicamente por telangiectasias, epistaxis a repetición, lesiones viscerales vasculares (malformaciones arteriovenosas) y usualmente con un antecedente familiar. Este trabajo reporta un caso clínico compatible con esta entidad que presentó complicaciones muy raras como lo son el absceso pulmonar y cerebral a la vez.
Palabras claves
Telangiectasia, síndrome de Osler-Weber-Rendu, hemorragia.
Abstract
Hereditary hemorrhagic telangiectasia (HHT) (also known as Osler Weber Rendu syndrome (OWRS)) is a rare dominant autosomal disorder whose frequency is between 1 per 1,331 people and 1 per 16,300 people depending on the population and its geographical location. There are no differences between genders. It is clinically characterized by telangiectasia, recurrent epistaxis, visceral vascular lesions (arteriovenous malformations - AVMs). Usually a person with HHT has a family history of the disorder. This paper reports a case that is clinically compatible with this rare entity and which presented simultaneous complications of pulmonary and cerebral abscesses.
Keywords
Telangiectasia, Osler Weber Rendu syndrome, hemorrhage.
CASO CLÍNICO
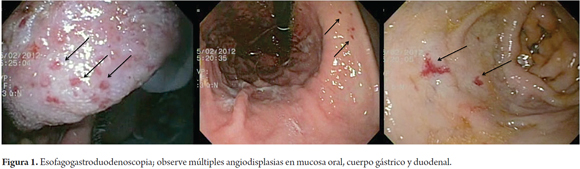
Paciente de 62 años masculino que consulta a nuestro hospital por presentar cuadro clínico de múltiples episodios de hematemesis hasta la anemización por lo que requirió soporte transfusional, se lleva a esofagogastroduodenoscopia en donde se informan angiodisplasias orofaríngeas, gástricas y duodenales. Como únicos antecedentes positivos alcoholismo pesado, niega sangrados o transfusiones, no procedimientos quirúrgicos previos no hay antecedente familiar relacionado con cuadro clínico actual (Figura 1).
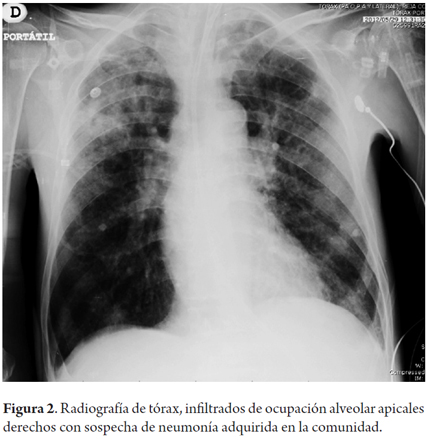
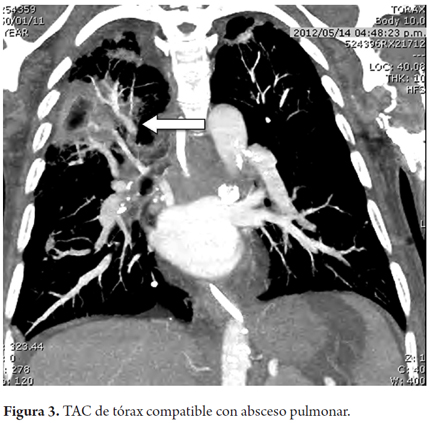
Al examen físico de ingreso el paciente se encontraba con deterioro cognitivo evidenciado por desorientación y con hemiparesia derecha, estaba postrado en cama desde hacía 10 días. Durante la evolución en el 3 día de hospitalización presenta picos febriles, disnea y deterioro progresivo, que lo lleva hasta la falla ventilatoria, lo que requirió soporte en unidad de cuidado intensivo. Se logra documentar la presencia de una neumonía (Figura 2) y posteriormente de un absceso pulmonar (Figura 3).
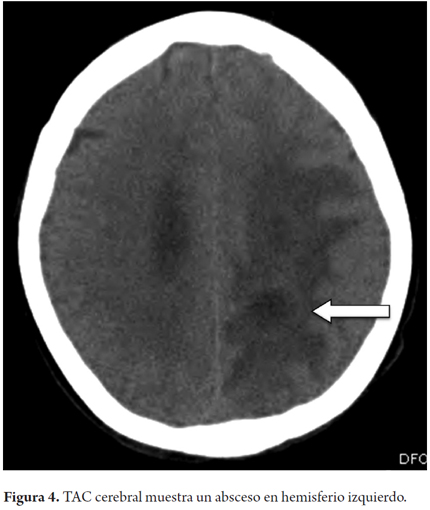
Dentro de los paraclínicos que se solicitaron de forma cronológica un ecocardiograma transesofágico con el fin de realizar diagnóstico diferencial de endocarditis infecciosa, en el cual se informa fracción de eyección del ventrículo izquierdo de 55% sin evidencia de valvulopatía o vegetaciones, recibe como parte de tratamiento múltiples esquemas de antibióticos de amplio espectro empírico (ampicilina sulbactam, piperacilina tazobactam y cefepime), pero el resultado fue deterioro progresivo pasando por sepsis severa y un estado vegetativo persistente, por lo cual se solicitó TC (tomografía computarizada) cerebral (Figura 4) y se documentó un proceso infeccioso intracerebral-absceso (que requirió ventriculostomía, catéter de drenaje de presión intracraneal); después, debido al soporte ventilatorio prolongado, requirió traqueostomía, pero continuó con una evolución tórpida y posteriormente fallece.
TELANGIECTASIA HEMORRÁGICA HEREDITARIA
La telangiectasia hemorrágica hereditaria o síndrome de Osler-Weber-Rendu descrita inicialmente por Sutton (1864) como un desorden caracterizado por epistaxis y una malformación del sistema vascular (1), después Benjamin Guy Babington lo describe como una epistaxis hereditaria haciendo referencia a su afectación familiar (2), luego Rendu (1896) lo describe como una entidad diferente a la hemofilia (3), Osler (1901) establece su carácter hereditario e informa de la presencia de malformaciones arteriovenosas viscerales (4); Weber (1907) publica la primera serie de casos (5). De estos tres últimos autores se tomó el epónimo que hasta ahora se reconoce y que tiene aceptación universal, aunque desde 1909 Hanes le otorga el término de telangiectasia hemorrágica hereditaria (THH), que para fines prácticos tiene la misma validez (6).
Desde el punto de vista epidemiológico la prevalencia de la THH no hay diferencia en cuanto a la afectación de género, por lo demás es variable y depende de la población y del área geográfica en donde se realice el estudio. En general puede variar de 1-3 casos por cada 500 a 5000 habitantes en el hemisferio oeste (7). Específicamente en EE.UU. Vermont, la prevalencia se estima en 1/16 500 personas, 1/2351 en Francia, 1/39 216 en el norte de Inglaterra y 1/100 000 en Alemania (8), la prevalencia más alta se ha encontrado en la región de Curasao y Bonaire, donde se ha reportado hasta en 1/1331 habitantes.
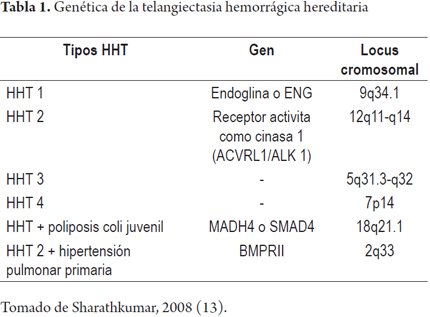
Se sabe que la THH tiene un rasgo autonómico dominante de penetrancia y expresión variable. Se han identificado al menos cinco mutaciones en genes que pueden causar HHT, estos genes codifican proteínas que modulan la actividad del factor de crecimiento transformante beta (TGF)-β superfamilia de señalización en las células endoteliales vasculares que regulan la proliferación celular, diferenciación, migración y formación de matriz extracelular. Según la afectación se reconocen 5 subtipos diferentes (Tabla 1), de los cuales las dos mutaciones más importantes por su frecuencia (>80%) son: en el cromosoma 9 (ENG, produce endoglina, OMIM # 187300) relacionado con HHT 1 y el cromosoma 12 (ACVRL1, proteína: receptor de activina-cinasa tipo 1, ALK-1, OMIM # 600376) relacionado con la HHT 2. Una pequeña proporción de los pacientes tienen HHT debido a una mutación en el gen MADH4 (producto de proteína: Smad4, OMIM # 175050) como parte de un síndrome de superposición de poliposis juvenil-HHT (10). Las mutaciones de ENG o ALK-1 llevan a la producción de proteínas inestables, con una actividad biológica insuficiente ya que no llegan a la superficie de la membrana o llevan a que la cantidad de moléculas de ENG o ALK-1 funcionalmente útiles sea baja, como consecuencia se presentan múltiples cambios como: disfunción de la división celular que limita la angiogénesis, adicionalmente la migración celular está alterada, lo que limita la configuración tridimensional en estructuras tubulares necesarias en la angiogénesis. Además existe una alteración del citoesqueleto, ya que la polimerización de los filamentos de actina cambia, lo que lleva a una disminución de la resistencia celular del endotelio vascular causando una lisis prematura de células endoteliales de los capilares, cuando estos son sometidos a estrés por un aumento de presión o trauma (11).
Actualmente se estudian las posibles funciones del factor de crecimiento vascular endotelial (VEGF) debido a que se han encontrado niveles elevados en HHT junto con TGF-β1, por el momento no se conoce si esta elevación es causa o consecuencia de la HHT (10).
Desde el punto de vista histopatológico se caracteriza por comprometer las paredes de arteriolas, vénulas y capilares, las cuales son más delgadas, produce una disminución en la elastofibrosis y en el grosor de la capa muscular lisa, lo que resulta en dilatación de la luz vascular y en malformaciones arteriovenosas que predispone a la ruptura. Existen tres patrones de displasia vascular en la HHT: malformaciones arteriovenosas (pulmón y cerebro, como pasó en nuestro paciente), telangiectasia (mucosa, dermis y compromiso visceral) y pseudoaneurismas. En todas se pueden observar telangiectasias de mayor o menor grado (7).
La clínica que caracteriza esta patología está íntimamente ligada a la patogenia, la formación anormal de los vasos y la hemorragia subsecuente son la base de todas las manifestaciones clínicas en HHT. Aunque el número y localización de las lesiones varía ampliamente, incluso dentro de una misma familia, la mayoría de las telangiectasias se encuentran en las mucosas oral, nasal y gastrointestinal (véase Figura 1), mientras que malformaciones arteriovenosas (MAV) se producen en pulmones, hígado y SNC en general; lo cual se presentó en nuestro paciente (véase Figura 2-4) manifestándose en sangrado digestivo, abscesos pulmonares y cerebrales. No es usual ver un caso de estos y para nuestro conocimiento es el primero reportado en Colombia.
Las lesiones telangiectásicas más pequeñas generalmente se presentan con síntomas de sangrado recurrente y anemia ferropénica secundaria, otros síntomas que se pueden ver en el contexto de pacientes con MAV más grandes son trombosis y embolismo (12,13). Las telangiectasias en la piel y las membranas mucosas, la presencia de epistaxis recurrente y una historia familiar positiva constituyen la tríada clásica de HHT. Se estima que 90% de los pacientes con HHT tienen epistaxis espontáneas y recurrentes, el 75% telangiectasias cutáneas; 30% afectación pulmonar o hepática (MAV), el 15% manifiesta sangrado gastrointestinal (SGI) y el 10%-20% tienen lesiones del SNC (13-14). La progresión sintomática es epistaxis, después se presentan malformaciones arteriovenosas pulmonares (MAVP), luego telangiectasias cutáneas y mucosas.
En cuanto a los abscesos cerebrales y al accidente cerebrovascular que ilustramos en el presente caso clínico, se presentan como causa directa de las MAVP con cortocircuitos de derecha a izquierda que causan trombosis, lo que genera una embolia paradójica, por lo que las MAVP deben ser consideradas como una causa probable en pacientes con abscesos cerebrales de etiología desconocida. Estas complicaciones pueden ser la primera manifestación de telangiectasia hemorrágica hereditaria (15-16). Sin embargo, un absceso cerebral es la complicación neurológica más grave de MAVP y ocurre en el 5% al 10% de los pacientes que presentan este tipo de malformaciones secundarias a HHT (17). Estos abscesos cerebrales tienen características clínicas radiológicas y bacteriológicas diferentes a los secundarios a otras entidades: son generalmente de localización en las capas superficiales de los lóbulos cerebrales (principalmente el lóbulo parietal), donde los microinfartos y el embolismo séptico suelen ocurrir. No están comúnmente asociados a infección por estafilococos sino a gérmenes anaerobios y aparecen más frecuentemente entre la tercera y la quinta décadas de la vida en relación al aumento del número y tamaño de MAVP (17,18). Desde el 2000 se han postulado unos criterios diagnósticos conocidos como los criterios de Curasao, los cuales también son descritos por las guías internacionales publicadas en 2009 (18,19).
1. Epistaxis espontánea recurrente, nocturnas
2. Telangiectasias mucocutáneas en sitios característicos:
- Labios
- Lengua
- Cavidad oral
- Nariz
- Pulpejos
3. Malformaciones arteriovenosas internas:
- Pulmonares
- Cerebrales
- Hepáticas
- Gastrointestinales
- SNC
4. Antecedente familiar de HHT de primera línea
Diagnóstico:
- Definida: 3 o más criterios
- Posible: 2 criterios
- Improbable: <2 criterios
El tratamiento en general de la HHT va orientado hacia la manifestación clínica predominante y su grado de severidad (10,20), las MAVP pueden ser tratadas exitosamente con embolización, lo cual no es una solución permanente y requiere vigilancia continua. La recomendación es la embolización de MAVP de 3 mm o más de diámetro, con el fin de disminuir las complicaciones tromboembólicas (8-17). Los pacientes con HHT requieren de profilaxis antibiótica previa a la realización de procedimientos odontológicos y procedimientos invasivos médicos como parte de la prevención de absceso cerebrales (21).
REFERENCIAS
1. Sutton HG. Epistaxis as an indication of impaired nutrition, and of degeneration of the vascular system. Med Mirror 1864;1:769. [ Links ]
2. Babington BG. Hereditary epistaxis. Lancet. 1865;2:362-3. [ Links ]
3. Rendu M. Epistaxis repetees chez un sujet porteur de petits angiomes cutanes et muquex. Bull Mem Soc Med Hop Paris. 1896;13:731-34. [ Links ]
4. Osler W. On a family form of recurring epistaxis associated with multiple telangiectases of the skin and mucous membranes. Bull Johns Hopkins Hosp. 1901;12:333-7. [ Links ]
5. Weber FP. Multiple hereditary developmental angiomata (telangiectases) of the skin and mucous membranes associated with recurring haemorrhages. Lancet. 1907;ii:160-2. [ Links ]
6. Hanes FM. Multiple hereditary telangiectasis causing hemorrhage (hereditary hemorrhagic telangiectasia). Bull Johns Hopkins Hosp. 1909;20:63-73. [ Links ]
7. Chuan-Qiang Q, Shou-Gang G, Yan H, Yu-Xin C. CT Manifestations of Osler-Weber-Rendu Syndrome in Liver: Report of Three Cases. J Clin Imaging Sci. 2012;2:26. [ Links ]
8. Guttmacher AE, Marchuk DA, White RI. Hereditary hemorrhagic telangiectasia. N Engl J Med. 1995;333(14):918-24. [ Links ]
9. Westermann CJJ, Rosina AF, De Vries V, de Coteau PA. The prevalence and manifestations of hereditary hemorrhagic telangiectasia in the Afro-Caribbean population of the Netherlands Antilles: a family screening. Am J Med Genet A. 2003;116A(4):324-8. [ Links ]
10. Govani FS, Shovlin CL. Hereditary haemorrhagic telangiectasia: a clinical and scientific review. Eur J Hum Genet. 2009;17(7):860-71. [ Links ]
11. Fernández-L A, Sanz-Rodriguez F, Blanco FJ, Bernabéu C, Botella LM. Hereditary hemorrhagic telangiectasia, a vascular dysplasia affecting the TGF-beta signaling pathway. Clin Med Res. 2006;4(1):66-78. [ Links ]
12. Kjeldsen AD, Møller TR, Brusgaard K, Vase P, Andersen PE. Clinical symptoms according to genotype amongst patients with hereditary haemorrhagic telangiectasia. J Intern Med. 2005;258(4):349-55. [ Links ]
13. Sharathkumar AA, Shapiro A. Hereditary haemorrhagic telangiectasia. Haemophilia. 2008;14(6):1269-80. [ Links ]
14. Begbie ME, Wallace GMF, Shovlin CL. Hereditary haemorrhagic telangiectasia (Osler-Weber-Rendu syn drome): a view from the 21st century. Postgrad Med J. 2003;79(927):18-24. [ Links ]
15. Maldonado LV, Absceso intramedular en paciente con enfermedad de Rendu-Osler-Weber. Medicina. 2007:714-16. [ Links ]
16. Grand'Maison A. Hereditary hemorrhagic telangiectasia. CMAJ. 2009;180(8):833-5. [ Links ]
17. Tabakow P, Jarmundowicz W, Czapiga B, Czapiga E. Brain abscess as the first clinical manifestation of multiple pulmonary arteriovenous malformations in a patient with hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber disease). Folia Neuropathol. 2005;43(1):41-4. [ Links ]
18. Mathis S, Dupuis-Girod S, Plauchu H, Giroud M, Barroso B, Ly KH, et al. Cerebral abscesses in hereditary haemorrhagic telangiectasia: a clinical and microbiological evaluation. Clin Neurol Neurosurg. 2012;114(3):235-40. [ Links ]
19. Shovlin CL, Guttmacher AE, Buscarini E, Faughnan ME, Hyland RH, Westermann CJ, et al. Diagnostic criteria for hereditary hemorrhagic telangiectasia (Rendu-Osler-Weber syndrome). Am J Med Genet. 2000;91(1):66-7. [ Links ]
20. Faughnan ME, Palda VA, Garcia-Tsao G, Geisthoff UW, McDonald J, Proctor DD, et al. International guidelines for the diagnosis and management of hereditary haemorrhagic telangiectasia. J Med Genet. 2011;48(2):73-87. [ Links ]
21. Olitsky SE. Hereditary hemorrhagic telangiectasia: diagnosis and management. Am Fam Physician. 2010;82(7):785-90. [ Links ]
