










Services on Demand
Journal
Article
 text in
text in  English (pdf)
English (pdf)
 Article in xml format
Article in xml format Article references
Article references
 Send this article by e-mail
Send this article by e-mailIndicators
-
 Cited by SciELO
Cited by SciELO -
 Access statistics
Access statistics
Related links
-
 Cited by Google
Cited by Google -
 Similars in
SciELO
Similars in
SciELO -
 Similars in Google
Similars in Google
Share

 Permalink
PermalinkRevista colombiana de Gastroenterología
Print version ISSN 0120-9957
Rev Col Gastroenterol vol.31 no.1 Bogotá Jan./Mar. 2016
Alcohol, Cirrhosis, and Genetic Predisposition
Mónica Marcela Gaviria C. Biol. MSc. (c) (1), Gonzalo Correa Arango MD. (1), María Cristina Navas N. MSc. PhD. (1)
(1) Gastrohepatology Group of the Faculty of Medicine at the Universidad de Antioquia in Medellín, Colombia.
Received: 15-12-14 Accepted: 26-01-16
Abstract
Liver cirrhosis is the third most common cause of death attributable to alcohol consumption throughout the world. More than 80% of chronic drinkers develop steatosis, and 20% to 40% develop other complications such as fibrosis, alcoholic hepatitis and cirrhosis. However, not everyone who chronically consumes alcohol develops cirrhosis. This is partly because of the genetic component of each individual. The level of activity of the enzymes that metabolize alcohol is influenced by polymorphisms of the genes that coding for these enzymes. This is one of the determining factors in the development of terminal liver disease in response to alcohol consumption. Among the enzymes involved in alcohol metabolism are alcohol dehydrogenase (ADH), cytochrome P450 2E1 (CYP2E1) and acetaldehyde dehydrogenase (ALDH). It has been reported that higher levels of activity of ADH and CYP2E1 and lower levels of activity of ALDH may be risk factors in some populations for accumulation of acetaldehyde which is toxic for the organism.
This literature review covers the most important aspects of alcohol metabolism including polymorphisms (genotypes) of enzymes involved in the metabolism of alcohol as a risk factor. A search through the PubMed database from 1990 to be held 2013 was conducted using the keywords alcoholic liver disease, ADH, ALDH, CYP2E1, and polymorphism.
Keywords
Alcoholic liver disease, ADH, ALDH, CYP2E1, polymorphism.
INTRODUCTION
Chronic consumption of alcohol causes 3.3 million deaths around the world each year. This is 5.9% of all deaths: 7.6% in men and 4.0% in women. It is a risk factor that increases general levels of morbidity and mortality in diseases around the world. In addition, it is credited with 5.1% of the global burden and disability. (1)
There is evidence of a causal relationship between alcohol and at least 200 diseases including gastritis, pancreatitis, cardiovascular disease, liver cirrhosis, hepatocellular carcinoma, and gastric cancer. Pathologies associated with chronic alcohol consumption are determined by the volume of alcohol consumed, the pattern of drinking and the quality of the alcohol consumed. (1,2) Alcohol metabolism is a complex process involving absorption, distribution and elimination. The liver metabolizes more than 90% of the alcohol in the body; alcohol becomes acetaldehyde as the result of the action of alcohol dehydrogenase (ADH), cytochrome P540-2E1 (CYP2E1) and catalase. Then acetaldehyde is converted to acetate and water by aldehyde dehydrogenase (ALDH). (3)
In vitro, some polymorphisms in genes encoding ADH, CYP2E1 and ALDH enzymes It have been shown to be associated with higher enzyme activity and the accumulation of metabolites such as acetaldehyde which has a toxic effect that produces liver damage tissue (4-6).
ALCOHOL METABOLISM
Ethanol is absorbed by the intestinal tract from which it is transported to the liver where 90% of alcohol is metabolized. Two percent to ten percent of alcohol is metabolized in the lungs and kidneys. (3,7)
There are three systems involved in alcohol metabolism within the liver (Figure 1). The most important is ADH which is found in the cytosol of hepatocytes. It catalyzes the formation of acetaldehyde by transferring hydrogen from the OH group to the nicotinamide cofactor of nicotinamide adenine dinucleotide (NAD+) to convert it into the reduced form of the molecule NADH. This is then converted through hydrogenation into NADPH. (7, 8)
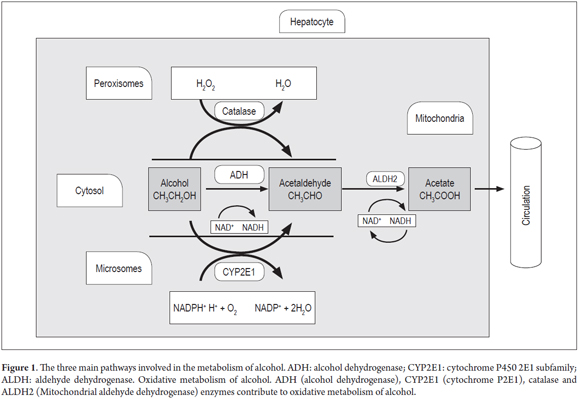
During the oxidation of acetaldehyde into acetate by the aldehyde dehydrogenase enzyme (ALDH), excess NADH is produced which alters the proportion of NADH to NAD. This effects the carbohydrate and lipid metabolism. NADH interferes with the transport of free fatty acids (FFA) and facilitates the formation of esterified fatty acids as the fatty acids react with the alcohol. This degrades polyunsaturated fatty acids by extracting hydrogen. The excess of NADH limits the availability of NAD which is necessary for the transport of FFA. (9) Acetate is incorporated into the Krebs cycle as acetyl coenzyme A (acetyl CoA), but if it is not transferred to the cycle, its accumulation may result in the production of ketone bodies leading to ketonuria and ketonemia. (7,9-13)
The second system is the microsomal ethanol oxidizing system (MEOS). It is an inducible system in which cytochrome P450 (CYP450) participates. Specifically CYP2E1 plays a major metabolic role in liver microsomes. Transcription of this gene is activated under conditions of high consumption of alcohol, and alcohol is metabolized to acetaldehyde using the phosphorylated NAD or reduced NADPH and oxygen (O2). This system metabolizes 3% to 8% of the alcohol. (14)
The third system operates in the peroxisomes of liver cells through catalase activity that metabolizes alcohol to acetaldehyde through peroxidation in the presence of hydrogen peroxide (H2O2) which is then transformed into water. This system metabolizes less than 2% of alcohol ingested. (3)
ALCOHOL AS A RISK FACTOR
Chronic alcohol consumption is a risk factor in 20% to 50% of liver cirrhosis cases worldwide. (1) In 2010, alcohol-attributable liver cirrhosis was responsible for 47.9% of deaths from liver disease. (15)
Eighty to ninety percent of chronic drinkers develop fatty liver disease and are at risk of developing complications such as steatohepatitis, fibrosis, alcoholic hepatitis, alcoholic cirrhosis and hepatocellular carcinoma. Alcoholic cirrhosis is characterized by fibrosis and distortion of the normal architecture of the liver (Figure 2). (7,16)
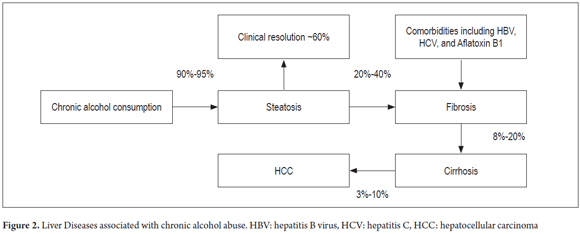
Cohort studies and case and control studies have established that consumption of more than 30 grams/day of ethanol increases the risk of cirrhosis eleven times (OR: 10.9, 95% CI: 3.6 to 33.5) and that consumption of 60 g/d of ethanol for five years increases the risk of hepatocellular carcinoma (HCC) seven times (OR: 7.0, 95% CI: 4.5 to 11.1). (17-19) The WHO estimates that each drink of beer, wine or distilled liquor contains approximately 10 grams of ethanol. In animal models, chronic alcohol consumption has been shown to affect the transport of vitamins in the small intestine. This was demonstrated in Sprague Dawley rats that were fed casein, vitamins, minerals, maltose and high concentrations of thiamine and alcohol for 6 to 8 weeks. Alcohol decreased thiamine transport and Na-K pump activity. (20)
A six-year study of baboons (Papio hamadryas) who ingested alcohol found that these primates developed a spectrum of liver diseases including steatosis, liver fibrosis and cirrhosis. Baboons who were not provided alcohol did not develop liver disease indicating that alcohol quickens the process of fibrosis and leads to the development of cirrhosis. (21-23)
Similarly, studies of humans to determine the risk of developing cirrhosis from alcohol have concluded that 50% of the phenotypic variation in terms of development of liver disease can be attributed to genetic factors. It has been observed that monozygotic twins more easily progress to alcoholic cirrhosis than do dizygotic twins. (24, 25)
An interaction between alcohol and HBV and HCV infections has also been demonstrated. Over a five-year follow-up period, the risk of developing HCC in those who consumed alcohol and who also had hepatitis B and C infections was twice that consumers of alcohol (60g/d) who did not have HBV or HCV. (17)
It has also been shown that patients who consume alcohol and have cirrhosis (diagnosed by histology and imaging) have a 24 times higher risk) of developing HCC than do patients without liver disease (OR: 23.8; 95% CI: 7.3 to 79. In addition, analysis of the synergistic effects of alcohol, obesity and smoking has shown that patients with this combination have a 7.4 times higher risk of developing HCC than do patients with only one of these risck factors (OR: 7.4, 95%: CI 2.1 to 14.6). (26)
ENZYMES INVOLVED IN ALCOHOL METABOLISM
Alcohol dehydrogenase (ADH)
This enzyme consists of two subunits which are encoded by the ADH1, ADH4, ADH5, ADH6 and ADH7 genes on the long arm of chromosome 4 (4q21-24) (27,28). These genes encode different subunits of the ADH of the liver. These subunits, α, β, γ, π, and χ, determine 12 isoenzymes (14). Isoenzymes are grouped into 5 classes (I-V) (Table 1). Classes I to III are present in the liver. (29) Class I is predominant with three isoenzymes. These isoenzymes, ADH1A, ADH1B, and ADH1C, exhibit great homology in 80% of the sequences. (30,31)
ADH enzymes are 40 kDa (kilo Dalton) dimers which contain zinc and are dependent on NAD. (32) They belong to the family of dehydrogenase and reductase enzymes that catalyze oxidation of alcohol to produce aldehyde or ketone. (33) Class I ADHs, which includes ADH1A, ADH1B, and ADH1C, is most common in the liver. Class II ADHs are present at low levels in the liver, and Class III ADHs are found in all tissues but are not very closely associated with alcohol metabolism. (33,34) ADH1A predominates in the first trimester of fetal development. After birth, expression of ADH1B and ADH1C increases. (35,36)
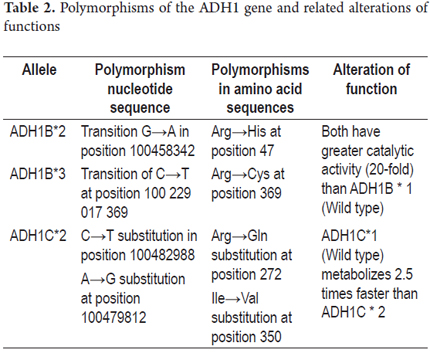
The ADH1B isoenzyme is expressed from the ADH1B*1 (β1β1) wild type allele which is present in more than 90% of Caucasian population. ADH1B*2 (β2β2) has been reported at high frequencies in Asian populations (~ 70%), and ADH1B*3 ( β3β3) is common in African populations (~ 16%). (37) The ADH1B and ADH1C isoenzymes present variations in their amino acid chain enzymes which generate greater catalytic activity (Table 2).
Aldehyde dehydrogenase (ALDH)
Aldehyde dehydrogenase (ALDH) is a super family of genes that encodes proteins that catalyze the conversion of substrates of aldehyde to carboxylates via NAD+. (38,39)
The cytosolic form is encoded by the ALDH1 gene which is located on the long arm of chromosome 9 in region 9q21.13. (40) The mitochondrial form is encoded by ALDH2 which is located in the long arm of chromosome 12 in the region 12q24.12. ALDH2 has greater importance because it metabolizes larger quantities of acetaldehyde. (14,40,41)
ALDH2 is a 54KDa mitochondrial enzyme with a tetrameric structure which catalyzes oxidation of more than 90% of the acetaldehyde produced by the oxidation of ethanol in liver detoxification. (14,42)
Two polymorphisms of the ALDH2 gene have been described: ALDH2*1 and ALDH2*2. The first is very active while the second has a mutation at amino acid 487 (substitution Glu→Lys). This mutation is associated with low specific activity which results in less efficient oxidation of acetaldehyde and produces an accumulation of this metabolite. (41,43) This causes various toxic effects in the liver including alteration of proteins in hepatocytes by binding the compound to the amino group of the proteins (Figure 1). (3,42)
The Caucasian population has two isoenzymes: ALDH1 and ALDH2. The ALDH2 allele described in Caucasian populations does not have a mutation associated with decreased ability to metabolize acetaldehyde.
Approximately 50% of the Japanese population loses ALDH2 isoenzyme activity because of the transition G/C→A/T in exon 12 with substitution of an amino acid Glu→Lys the at position 14 of the terminal COOH. This generates a defective protein in the catalytic site and thus decreases metabolic activity. (38)
Cytochrome P450 (CYP2E1)
The CYP450 familys main function is to metabolize various xenobiotic substances that enter the body. These include medications, drugs and alcohol. There are 44 subfamilies of which subfamily IIE is the most important in the metabolism of alcohol. Subfamily IIE of CYP2E1 is encoded by a gene located on chromosome 10 in the region 10q26.3. (44)
The CYP2E1 gene encodes a 56.9 KD enzyme that is capable of metabolizing drugs, hormones and xenobiotic toxins such as ethanol. The enzyme has a redox function and is localized in the membrane of the endoplasmic reticulum. (6,44-46)
Transcriptional induction occurs at high levels of ethanol, so in situations of chronic drinking there is a high rate of transcription. CYP2E1 participates in the microsomal ethanol oxidation system (MEOS) as the reticle (Figure 1) whose function is to metabolize ethanol to acetaldehyde. During this process ROS is produced. (47,48)
A relationship between the level of expression of CYP450 and the amount of DNA adducts has been reported. DNA adducts are formed through binding of compounds such as acetaldehyde and modified bases, which are known for their ability to induce mutations in the DNA that facilitates activation of oncogenes, through inactivation of tumor suppressor genes, and through immune response due to recognition of proteins bound to foreign structures by the immune system. (6,49)
Among the polymorphisms of CYP2E1which have been studied, CYP2E1*5B corresponds to a change of sequence of regulatory region 5`of the C-1053T gene. This change is associated with a higher rate of transcription and enzymatic activity and has been reported with increased risk of liver diseases associated with alcohol consumption. (44)
Catalase
The gene encoding catalase is located on chromosome 11 in the 11p13 region. (50) Catalase is an oligomeric protein with four 60kDa subunits. (14) The catalase system (Figure 1) is located in the peroxisomes. Its main function is to regulate levels of hydrogen peroxide and peroxidation of ethanol to acetaldehyde in the presence of H2O2 (51).
Catalase has several isoforms. Type I is characterized by a change in position of G→A in position 5 of intron 4. Type II has a deletion in position 358 of T in exon 4, and Type III has an insert in position 138GA of exon 2. (52-54) These mutations are associated with decreased catalytic activity and therefore with decreased metabolism (<10%) of hydrogen peroxide. This causes a condition called acatalasemia which is an autosomal recessive syndrome in which the catalase activity in erythrocytes is decreased. (54)
PATHOPHYSIOLOGY OF ALCOHOL AND CIRRHOSIS
Acetaldehyde is a metabolite of alcohol that, when it accumulates, produces liver damage as the result of its ability to generate formation of DNA adducts such as sites without purine or pyrimidine (Figure 3). Similarly, alcohol metabolism by CYP2E1 enzyme generates reactive oxygen species including hydrogen peroxide (H2O2), superoxide and hydroxyl radicals which have important roles in causing damage to DNA and in fatty acid oxidation. (6)
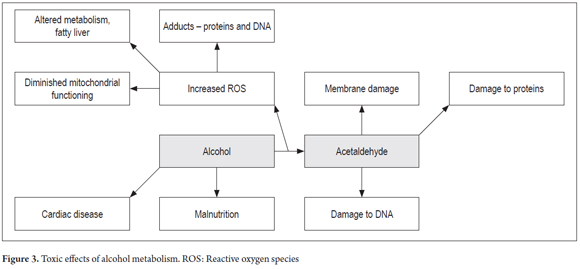
Acetaldehyde has toxic effects such as damage to mitochondria due to alterations in the cell membrane, DNA damage, reduction of oxygen utilization, cell death due to decreases in enzyme activity of the proteins that degrade reactive oxygen species (ROS) such as glutathione s transferase (GST) and lipid peroxidation. (7,9-13)
Due to lipid peroxidation resulting from alcohol abuse, hepatic steatosis develops through accumulation of fat in vacuoles of different sizes in the cytoplasm of hepatocytes. In turn, macro-vacuoles may displace the nucleus to the periphery of the cell. Inflammation caused by infiltration of polymorphonuclear leukocytes can worsen steatosis leading to the next stage which is called steatohepatitis. (15,55) In hepatic steatosis, alterations occur in the fabric structure. They are usually accumulations of free fatty acids located in the center-lobular areas. They appear droplets of fat together with the cellular infiltrates mentioned above. Conversion of acetyl CoA to fatty acids and lipogenesis also increase. (16,55,56)
Alcoholic hepatitis is another manifestation of alcoholic liver disease that is characterized by cell necrosis resulting from infiltration of polymorphonuclear leukocytes and hepatocyte degeneration localized in the center lobular. (56)
The death of hepatocytes induced by ethanol consumption can trigger liver fibrosis and produce a fibrotic regeneration with accumulation of type I collagen. This occurs because of increased synthesis of type I collagen by hepatic stellate cells (HSCs) and increased production of the extracellular matrix protein called osteopontin. HSCs can be activated by hepatocytes with cellular damage and can simultaneously increase synthesis of collagenase inhibitors, and activate Kupffer cells and neutrophils. This leads to chronic liver damage resulting from increased amounts of growth factor, cytokines, chemokines, and free radicals that reactivate HSCs and increase their effects leading to greater liver complications such as cirrhosis. (16,57)
POLYMORPHISMS AND GENETIC SUSCEPTIBILITY TO DEVELOPMENT OF CIRRHOSIS
In 1965, Von Wartburg, Papenberg and Aebi reported that certain individuals had an atypical form of the alcohol dehydrogenase enzyme, (58) and Greenfield and Pietruzko reported two ALDH isoenzymes that had different metabolic activities. (59)
Decreased metabolic activity of ADH was demonstrated in Caucasian populations. An analysis of groups of Caucasian patients with alcoholic cirrhosis, non-alcoholic cirrhosis, chronic drinkers without liver disease and moderate drinkers without liver disease found that the activity of ADH is lower in patients with alcoholic cirrhosis. This activity was also lower in cirrhotic patients with polymorphism ADH1B1 than in cirrhotic patients with polymorphism ADH1B2. In addition, patients with chronic alcohol consumption had lower levels of ALDH activity which made it evident that varying levels of metabolic efficiency are associated with variation of gene expression. (29)
Among Japanese people, the ALDH gene mutation which encodes an inactive form of the enzyme causes a response called Oriental flushing response. Patients with this mutation have headaches, nausea and dizziness and must reduce alcohol consumption. (60) However, studies are still inconclusive because increased activity of the ADH (ADH1*B2) enzyme and decreased activity of the ALDH (ALDH2*2) enzyme have also been described. This generates increased formation of acetaldehyde, lower efficiency of oxidation which causes damage to cellular tissue and toxicity. This has also been confirmed in vitro. (61)
It has also been proposed that the risk of developing cirrhosis may be related to interactions between genes encoding metabolic enzymes. In other words, when combined with a slow metabolism, expression of ALDH2*2, the ALDH gene with low metabolic activity, and expression of the ADH1B*2 and CYP2E1*5B variants that cause increased production of acetaldehyde, could be responsible for damage to cells. (62) In contrast to these results, another study has found increased incidence of the ALDH2 * 1 variant in patients with alcoholic cirrhosis. (63) Table 3 summarizes some of the studies of polymorphisms of ADH, ALDH and CYP2E1 genes that have been linked to the development of alcoholic liver disease.

CONCLUSION
In conclusion, ethanol is an important cellular toxin which generates various substances such as acetaldehyde and ROS which directly damage cells. Ethanol activates hepatic stellate cells to produce abnormally large amounts of collagen production and results in changes in liver structure which cause fibrosis. This can progress to cirrhosis. There is evidence that combinations of polymorphisms in genes encoding enzymes that metabolize alcohol, including ADH1B*2, ADH1C*1, ALDH2*2 and CYP2E1*5, increase susceptibility to development of liver disease through accumulation of acetaldehyde in the organism.
Our study has shown that there is a need for additional studies on the toxicity of alcohol in the body that take into account genotypes for enzymes that metabolize ethanol and which contribute to the prognosis of the ability of an individual to remove ethanol from the body.
Funding
This publication is part of Project 11556935008 funded by the Administrative Department of Science, Technology and Innovation (COLCIENCIAS) and the Sustainability Project of the Office of the Vice-Rector for Research of the Universidad de Antioquia.
REFERENCES
1. Organization WH. Global status report on alcohol and health. Geneva: World Health Organization 2014 (citado 9 de septiembre de 2014). Available from: http://www.who.int/iris/bitstream/10665/112736/1/9789240692763_eng.pdf?ua=1 Available from: http://www.who.int/substance_abuse/publications/global_alcohol_report/en/ [ Links ]
2. Rehm J, Mathers C, Popova S, Thavorncharoensap M, Teerawattananon Y, Patra J. Global burden of disease and injury and economic cost attributable to alcohol use and alcohol-use disorders. Lancet. 2009;373(9682):2223-33. [ Links ]
3. Zakhari S. Overview: How is alcohol metabolized by the body? Alcohol Res Health. 2006;29(4):245-54. [ Links ]
4. Homann N, Stickel F, Konig IR, Jacobs A, Junghanns K, Benesova M, et al. Alcohol dehydrogenase 1C*1 allele is a genetic marker for alcohol-associated cancer in heavy drinkers. Int J Cancer. 2006;118(8):1998-2002. [ Links ]
5. Yokoyama A, Mizukami T, Matsui T, Yokoyama T, Kimura M, Matsushita S, et al. Genetic Polymorphisms of Alcohol Dehydrogenase-1B and Aldehyde Dehydrogenase-2 and Liver Cirrhosis, Chronic Calcific Pancreatitis, Diabetes Mellitus, and Hypertension Among Japanese Alcoholic Men. Alcohol Clin Exp Res. 2013;37(8):1391-401. [ Links ]
6. Wang Y, Millonig G, Nair J, Patsenker E, Stickel F, Mueller S, et al. Ethanol-induced cytochrome P4502E1 causes carcinogenic etheno-DNA lesions in alcoholic liver disease. Hepatology. 2009;50(2):453-61. [ Links ]
7. Baraona E, Lieber CS. Effects of ethanol on lipid metabolism. J Lipid Res. 1979;20(3):289-315. [ Links ]
8. Guynn RW, Pieklik JR. Dependence on dose of the acute effects of ethanol on liver metabolism in vivo. J Clin In est. 1975;56(6):1411-9. [ Links ]
9. Lundquist F, Tygstrup N, Winkler K, Jensen KB. Glycerol metabolism in the human liver: Inhibition by ethanol. Science. 1965;150(3696):616-7. [ Links ]
10. Korsten MA, Matsuzaki S, Feinman L, Lieber CS. High blood acetaldehyde levels after ethanol administration. Difference between alcoholic and nonalcoholic subjects. N Engl J Med. 1975;292(8):386-9. [ Links ]
11. Minana JB, Gomez-Cambronero L, Lloret A, Pallardo FV, Del Olmo J, Escudero A, et al. Mitochondrial oxidative stress and CD95 ligand: A dual mechanism for hepatocyte apoptosis in chronic alcoholism. Hepatology. 2002;35(5):1205-14. [ Links ]
12. Lefevre AF, DeCarli LM, Lieber CS. Effect of ethanol on cholesterol and bile acid metabolism. J Lipid Res. 1972;13(1):48-55. [ Links ]
13. Crouse JR, Gerson CD, DeCarli LM, Lieber CS. Role of acetate in the reduction of plasma free fatty acids produced by ethanol in man. J Lipid Res. 1968;9(4):509-12. [ Links ]
14. Riveros-Rosas H, Julian-Sanchez A, Pina E. Enzymology of ethanol and acetaldehyde metabolism in mammals. Arch Med Res. 1997;28(4):453-71. [ Links ]
15. Rehm J, Shield K. Global Alcohol-Attributable deaths From Cancer, Liver Cirrhosis, and Injury in 2010 Alcohol Res Curr Rev. 2014;35(2):10. [ Links ]
16. Orman ES, Odena G, Bataller R. Alcoholic liver disease: Pathogenesis, management, and novel targets for therapy. J Gastroenterol Hepatol. 2013;28 Suppl 1:77-84. [ Links ]
17. Donato F, Tagger A, Gelatti U, Parrinello G, Boffetta P, Albertini A, et al. Alcohol and hepatocellular carcinoma: the effect of lifetime intake and hepatitis virus infections in men and women. Am J Epidemiol. 2002;155(4):323-31. [ Links ]
18. Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut. 1997;41(6):845-50. [ Links ]
19. Frezza M, di Padova C, Pozzato G, Terpin M, Baraona E, Lieber CS. High blood alcohol levels in women. The role of decreased gastric alcohol. N Engl J Med. 1990;322(2):95-9. [ Links ]
20. Hoyumpa AM, Jr., Nichols S, Henderson GI, Schenker S. Intestinal thiamin transport: Effect of chronic ethanol administration in rats. Am J Clin Nutr. 1978;31(6):938-45. [ Links ]
21. Lieber CS, DeCarli LM. An experimental model of alcohol feeding and liver injury in the baboon. J Med Primatol. 1974;3(3):153-63. [ Links ]
22. Lieber CS, Robins SJ, Li J, DeCarli LM, Mak KM, Fasulo JM, et al. Phosphatidylcholine protects against fibrosis and cirrhosis in the baboon. Gastroenterology. 1994;106(1):152-9. [ Links ]
23. Lieber CS, DeCarli LM, Mak KM, Kim CI, Leo MA. Attenuation of alcohol-induced hepatic fibrosis by polyunsaturated lecithin. Hepatology. 1990;12(6):1390-8. [ Links ]
24. Hrubec Z, Omenn GS. Evidence of genetic predisposition to alcoholic cirrhosis and psychosis: twin concordances for alcoholism and its biological end points by zygosity among male veterans. Alcohol Clin Exp Res. 1981;5(2):207-15. [ Links ]
25. Reed T, Page WF, Viken RJ, Christian JC. Genetic Predisposition to Organ-Specific Endpoints of Alcoholism. Alcohol Clin Exp Res. 1996;20(9):1528-33. [ Links ]
26. Marrero JA, Fontana RJ, Fu S, Conjeevaram HS, Su GL, Lok AS. Alcohol, tobacco and obesity are synergistic risk factors for hepatocellular carcinoma. J Hepatol. 2005;42(2):218-24. [ Links ]
27. Osier MV, Pakstis AJ, Soodyall H, Comas D, Goldman D, Odunsi A, et al. A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet. 2002;71(1):84-99. [ Links ]
28. Hoog JO, Ostberg LJ. Mammalian alcohol dehydrogenases: A comparative investigation at gene and protein levels. Chem Biol Interact. 2011;191(1-3):2-7. [ Links ]
29. Poupon RE, Nalpas B, Coutelle C, Fleury B, Couzigou P, Higueret D. Polymorphism of alcohol dehydrogenase, alcohol and aldehyde dehydrogenase. Hepatology. 1992;15(6):1017-22. [ Links ]
30. Carrigan MA, Uryasev O, Davis RP, Zhai L, Hurley TD, Benner SA. The natural history of class I primate alcohol dehydrogenases includes gene duplication, gene loss, and gene conversion. PLoS One. 2012;7(7):e41175. [ Links ]
31. Stewart MJ, McBride MS, Winter LA, Duester G. Promoters for the human alcohol dehydrogenase genes ADH1, ADH2, and ADH3: Interaction of CCAAT/enhancer-binding protein with elements flanking the ADH2 TATA box. Gene. 1990;90(2):271-9. [ Links ]
32. Vallee BL, Bazzone TJ. Isozymes of human liver alcohol dehydrogenase. Isozymes Curr Top Biol Med Res. 1983;8:219-44. [ Links ]
33. Duester G, Farres J, Felder MR, Holmes RS, Hoog JO, Pares X, et al. Recommended nomenclature for the vertebrate alcohol dehydrogenase gene family. Biochem Pharmacol. 1999;58(3):389-95. [ Links ]
34. Persson B, Hedlund J, Jornvall H. Medium- and short-chain dehydrogenase/reductase gene and protein families. Cell Mol Life Sci. 2008;65(24):3879-94. [ Links ]
35. Smith M, Hopkinson DA, Harris H. Alcohol dehydrogenase isozymes in adult human stomach and liver: evidence for activity of the ADH 3 locus. Ann Hum Genet. 1972;35(3):243-53. [ Links ]
36. Smith M, Hopkinson DA, Harris H. Developmental changes and polymorphism in human alcohol dehydrogenase. Ann Hum Genet. 1971;34(3):251-71. [ Links ]
37. Birley AJ, James MR, Dickson PA, Montgomery GW, Heath AC, Martin NG, et al. ADH single nucleotide polymorphism associations with alcohol metabolism in vivo. Hum Mol Genet. 2009;18(8):1533-42. [ Links ]
38. Yoshida A, Shibuya A. Polymorphisms of Alcohol and Aldehyde Dehydrogenases and Their Significance for Alcohol Liver Diseases. In: Watson R, editor. Liver Pathology and Alcohol. Drug and Alcohol Abuse Reviews. 2: Humana Press; 1991. p. 441-66. [ Links ]
39. Black WJ, Stagos D, Marchitti SA, Nebert DW, Tipton KF, Bairoch A, et al. Human aldehyde dehydrogenase genes: Alternatively spliced transcriptional. Pharmacogenet Genomics. 2009;19(11):893-902. [ Links ]
40. Hsu LC, Yoshida A, Mohandas T. Chromosomal assignment of the genes for human aldehyde dehydrogenase-1 and aldehyde dehydrogenase-2. Am J Hum Genet. 1986;38(5):641-8. [ Links ]
41. Hsu LC, Bendel RE, Yoshida A. Genomic structure of the human mitochondrial aldehyde dehydrogenase gene. Genomics. 1988;2(1):57-65. [ Links ]
42. Crabb DW, Matsumoto M, Chang D, You M. Overview of the role of alcohol dehydrogenase and aldehyde dehydrogenase and their variants in the genesis of alcohol-related pathology. Proc Nutr Soc. 2004;63(1):49-63. [ Links ]
43. Farres J, Wang X, Takahashi K, Cunningham SJ, Wang TT, Weiner H. Effects of changing glutamate 487 to lysine in rat and human liver mitochondrial aldehyde dehydrogenase. A model to study human (Oriental type) class 2 aldehyde dehydrogenase. J Biol Chem. 1994;269(19):13854-60. [ Links ]
44. Umeno M, McBride OW, Yang CS, Gelboin HV, Gonzalez FJ. Human ethanol-inducible P450IIE1: Complete gene sequence, promoter characterization, chromosome mapping, and cDNA-directed expression. Biochemistry. 1988;27(25):9006-13. [ Links ]
45. Ravindranath V, Anandatheerthavarada HK, Shankar SK. NADPH cytochrome P-450 reductase in rat, mouse and human brain. Biochem Pharmacol. 1990;39(6):1013-8. [ Links ]
46. Shen Z, Wells RL, Liu J, Elkind MM. Identification of a cytochrome P450 gene by reverse transcription--PCR using. Proc Natl Acad Sci USA. 1993;90(24):11483-7. [ Links ]
47. Lieber CS. Ethanol metabolism, cirrhosis and alcoholism. Clin Chim Acta. 1997;257(1):59-84. [ Links ]
48. Hu Y, Ingelman-Sundberg M, Lindros KO. Induction mechanisms of cytochrome P450 2E1 in liver: Interplay between ethanol treatment and starvation. Biochem Pharmacol. 1995;50(2):155-61. [ Links ]
49. Albano E, Vidali M. Immune mechanisms in alcoholic liver disease. Genes Nutr. 2010;5(2):141-7. [ Links ]
50. Bell GI, Najarian RC, Mullenbach GT, Hallewell RA. cDNA sequence coding for human kidney catalase. Nucleic Acids Res. 1986;14(13):5561-2. [ Links ]
51. Koechling UM, Amit Z. Relationship between blood catalase activity and drinking history in a human. Alcohol. 1992;27(2):181-8. [ Links ]
52. Wen JK, Osumi T, Hashimoto T, Ogata M. Molecular analysis of human acatalasemia. Identification of a splicing mutation. J Mol Biol. 1990;211(2):383-93. [ Links ]
53. Kishimoto Y, Murakami Y, Hayashi K, Takahara S, Sugimura T, Sekiya T. Detection of a common mutation of the catalase gene in Japanese acatalasemic patients. Hum Genet. 1992;88(5):487-90. [ Links ]
54. Góth L, Shemirani A, Kalmár T. A Novel Catalase Mutation (a GA Insertion) Causes the Hungarian Type of Acatalasemia. Blood Cell Mol Dis. 2000;26(2):151-4. [ Links ]
55. Farfan Labonne BE, Gutierrez M, Gomez-Quiroz LE, Konigsberg Fainstein M, Bucio L, Souza V, et al. Acetaldehyde-induced mitochondrial dysfunction sensitizes hepatocytes to oxidative damage. Cell Biol Toxicol. 2009;25(6):599-609. [ Links ]
56. Pares A, Caballería J. Patología orgánica. Adicciones. 2002;14:17. [ Links ]
57. Gao B, Bataller R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology. 2011; 141(5): 1572-85. [ Links ]
58. Von Wartbung JP, Papenberg J, Aebi H. An atypical human alcohol dehydrogenase. Can J Biochem. 1965:43,889. [ Links ]
59. Greenfield NJ, Pietruszko R. Two aldehyde dehydrogenases from human liver. Isolation via affinity chromatography and characterization of the isozymes. Biochim Biophys Acta. 1977:483:35-45. [ Links ]
60. Harada S, Misawa S, Agarwal DP, Goedde HW. Liver alcohol dehydrogenase and aldehyde dehydrogenase in the Japanese: Isozyme. Am J Hum Genet. 1980;32(1):8-15. [ Links ]
61. Khan AJ, Husain Q, Choudhuri G, Parmar D. Association of polymorphism in alcohol dehydrogenase and interaction with other. Drug Alcohol Depen. 2010;109(1-3):190-7. [ Links ]
62. Tanaka F, Shiratori Y, Yokosuka O, Imazeki F, Tsukada Y, Omata M. High incidence of ADH2*1/ALDH2*1 genes among Japanese alcohol dependents and patients with alcoholic liver disease. Hepatology. 1996;23:234-9. [ Links ]
63. Muramatsu T, Wang ZC, Fang YR, Hu KB, Yan H, Yamada K, et al. Alcohol and aldehyde dehydrogenase genotypes and drinking behavior of Chinese living in Shanghai. Hum Genet. 1995;96(2):151-4. [ Links ]
